Downloaded 638 times





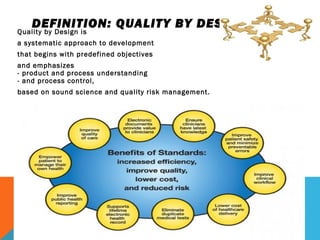
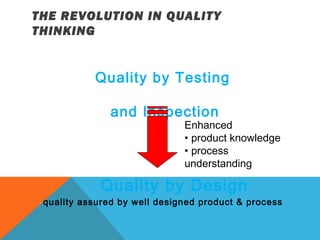




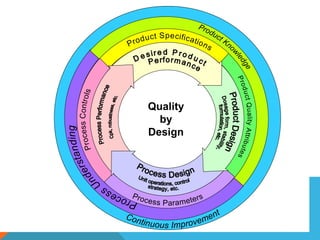



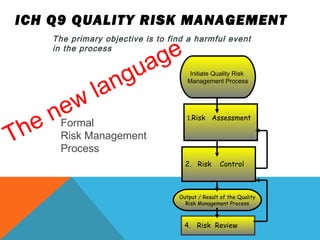

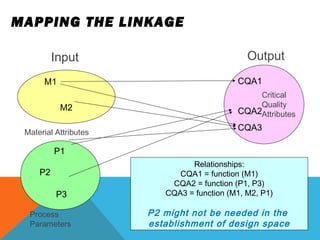
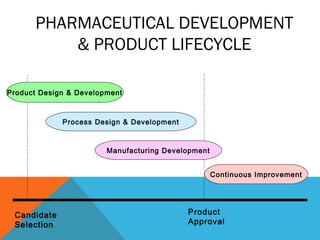
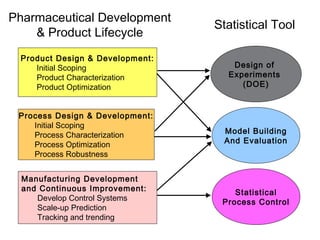















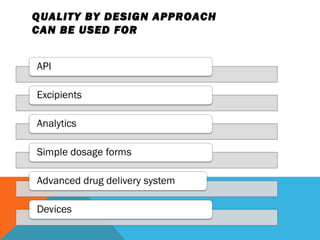
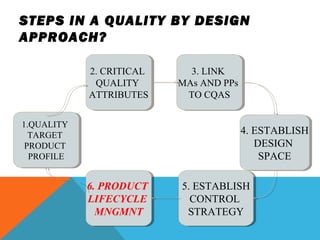



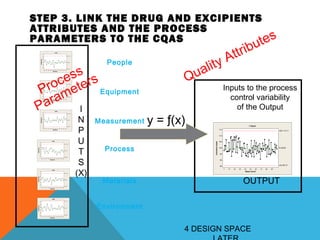




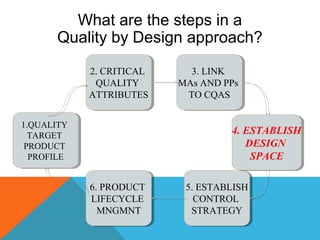
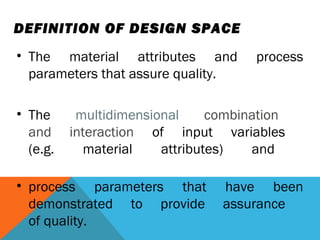
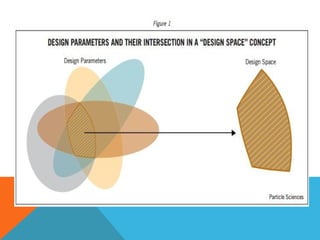
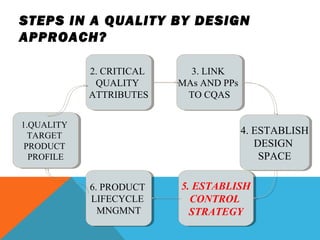
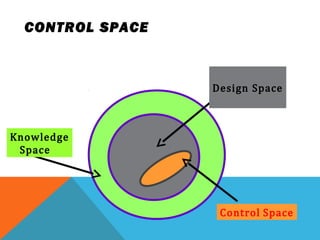

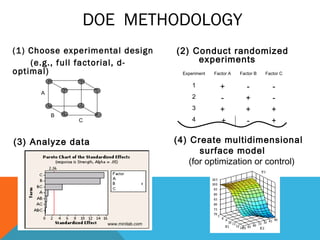

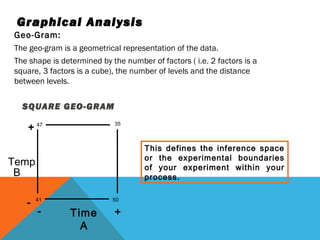
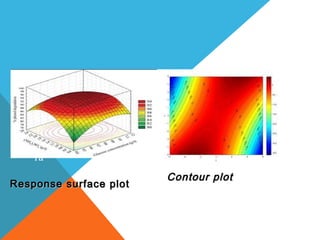
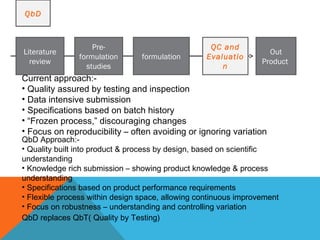
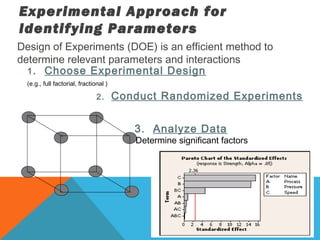
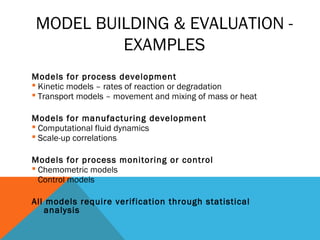





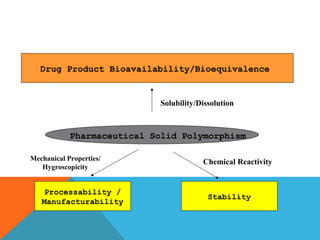
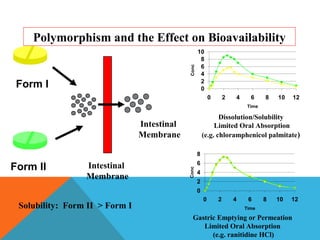
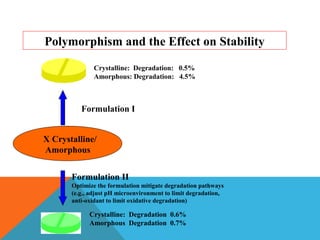
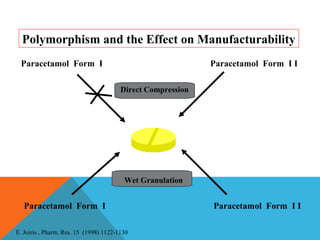
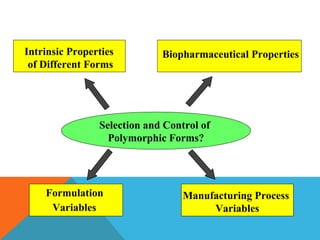
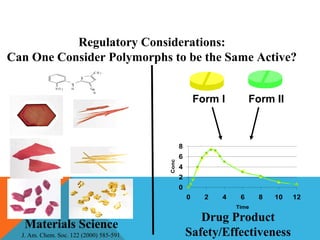


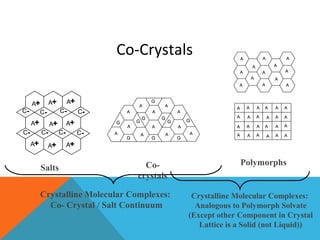
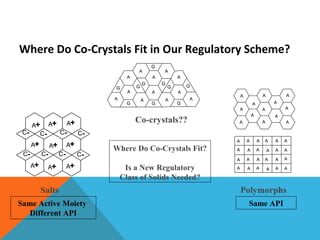


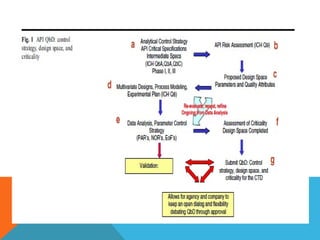

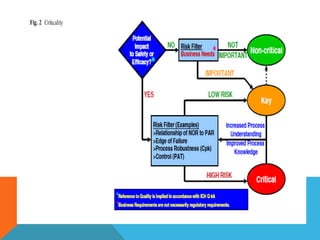
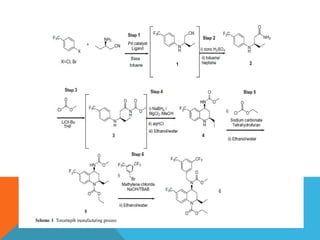
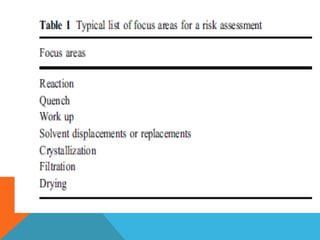
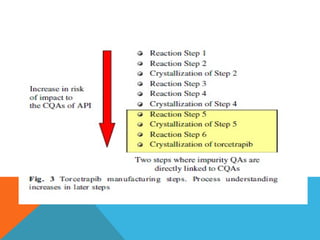
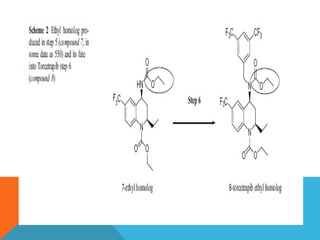
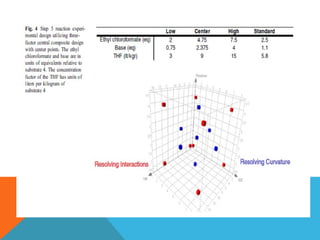
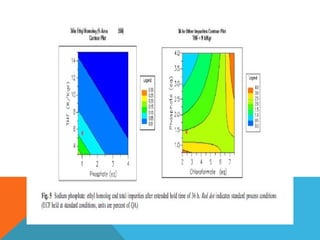
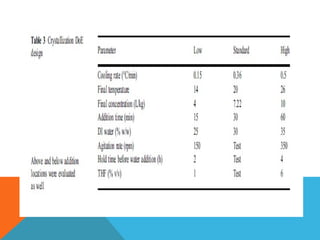
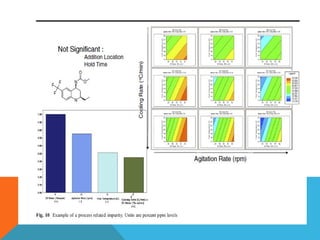
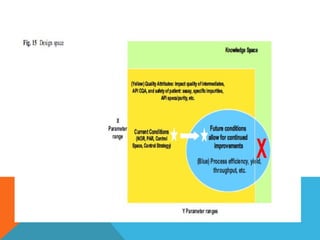
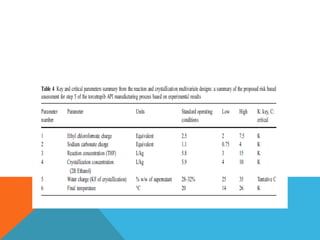

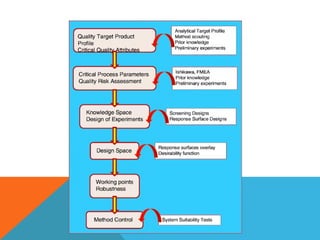









This document discusses Quality by Design (QbD) in API manufacturing. It begins by quoting Joseph M. Juran that quality must be built in by design, not tested in. It then defines QbD as a systematic approach that emphasizes product and process understanding and control based on science and risk management. The document outlines some of the key aspects of QbD like design space, quality risk management, and using tools like design of experiments. It compares the traditional and QbD approaches and discusses some of the pros and cons of QbD.
























































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)


















