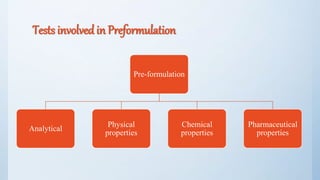
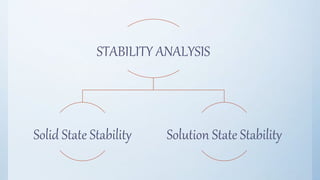
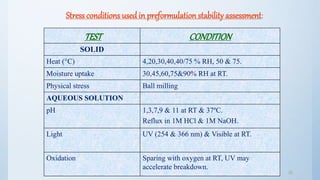
The document discusses preformulation studies that are conducted prior to developing a dosage form for a new drug. The objectives are to determine the drug's physicochemical properties, solubility, stability, and compatibility with excipients. Key tests include solubility studies under various conditions, stability studies of solid and liquid states under stress conditions like heat, light and pH, and chemical characterization of properties like oxidation and hydrolysis. The outcomes aim to develop a formulation that safely delivers the drug to the site of action as intended.











![AQUEOUS SOLUBILITY
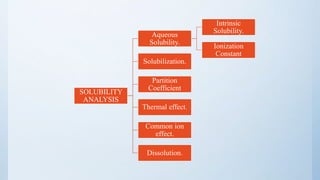
There are two fundamental properties mandatory for a new compound.
INTRINSIC SOLUBILITY (CO) :-
S = So {1 + (K1 / [H+])} ------ for weak acids.
S = So {1 + ([H+] / K2)} ------ for weak bases.
where, S = Solubility at a given pH.
So = Intrinsic solubility of the neutral form.
K1 = Dissociation constant of weak acid.
K2 = Dissociation constant of weak base.
14](https://image.slidesharecdn.com/preformulationstudiespart2mz-180120053417/85/Preformulation-studies-part-2-mz-14-320.jpg)

![ionization constant:-
75 % of all drugs are weak bases,
20 % are weak acids and only,
5 % are nonionic amphoteric or alcohol.
Henderson-Hasselbalch equation:-
pH = pKa + log [ionized form] / [unionized form] --- for acids.
pH = pKa + log [unionized form] / [ionized form] --- for bases.
16](https://image.slidesharecdn.com/preformulationstudiespart2mz-180120053417/85/Preformulation-studies-part-2-mz-16-320.jpg)

















![3636
Prevention of oxidation:-
Reducing oxygen content.
Storage in a dark & cool condition.
Addition of chelating agent. [E.g.. EDTA, Citric acid, Tartaric acid].
Adjustment of pH.
Changing solvent. [E.g.. Aldehydes, ethers, ketones may influence
free radical reaction].
Addition of an antioxidant.
• Reducing agent.
• Chain inhibitors of radical induced decomposition](https://image.slidesharecdn.com/preformulationstudiespart2mz-180120053417/85/Preformulation-studies-part-2-mz-36-320.jpg)









![4646
Avoiding sunbath.
E.g.. Sparfloxacin.
Photostabilizer [Light absorber].
Colorant Curcumin, Azorubine.
Pigments Iron oxide, Titanium dioxide.
Coating:
Pigments like TiO2(IN NIFEDIPINE) / ZnO.
E.g.. Photostabilization of Sulphasomidine Tab. by film
coating containing U.V. absorber (Oxybenzone) to protect
color & photolytic degradation.](https://image.slidesharecdn.com/preformulationstudiespart2mz-180120053417/85/Preformulation-studies-part-2-mz-46-320.jpg)

![4848
POLYMERIZATION
It is a continuous reaction between molecules.
More than one monomer reacts to form a polymer.
Eg. Darkening of glucose solution is attributed to
polymerization of breakdown product [5- (hydroxyl methyl)
furfural].
Eg. Shellac on aging undergoes polymerization & hence
prolongs disintegration time &dissolution time.](https://image.slidesharecdn.com/preformulationstudiespart2mz-180120053417/85/Preformulation-studies-part-2-mz-48-320.jpg)








































































