Downloaded 1,127 times







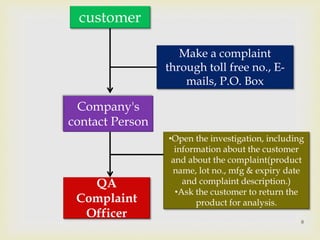









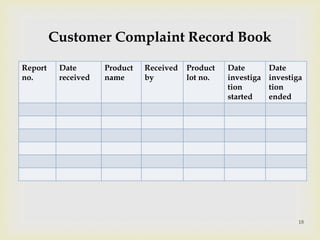

























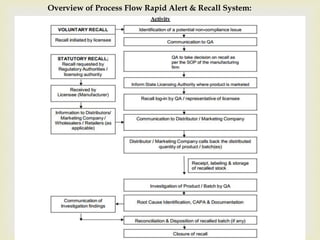










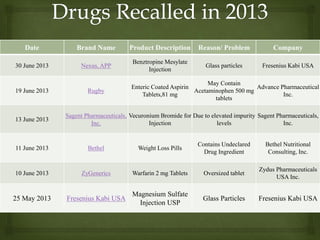
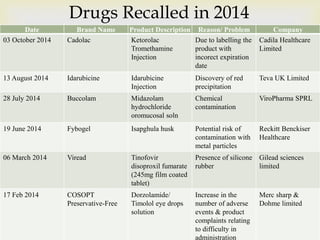



The document discusses product complaints and recalls in the pharmaceutical industry. It defines a complaint as customer dissatisfaction about a product and outlines four types of complaints. It details the four-step process of handling complaints, which includes receiving, investigating, implementing corrective actions, and reporting. The document also defines a recall as removing a product from distribution due to quality, safety or efficacy issues. It describes the reasons and types of recalls, as well as the classification, levels and timelines involved in an effective recall system.























































































