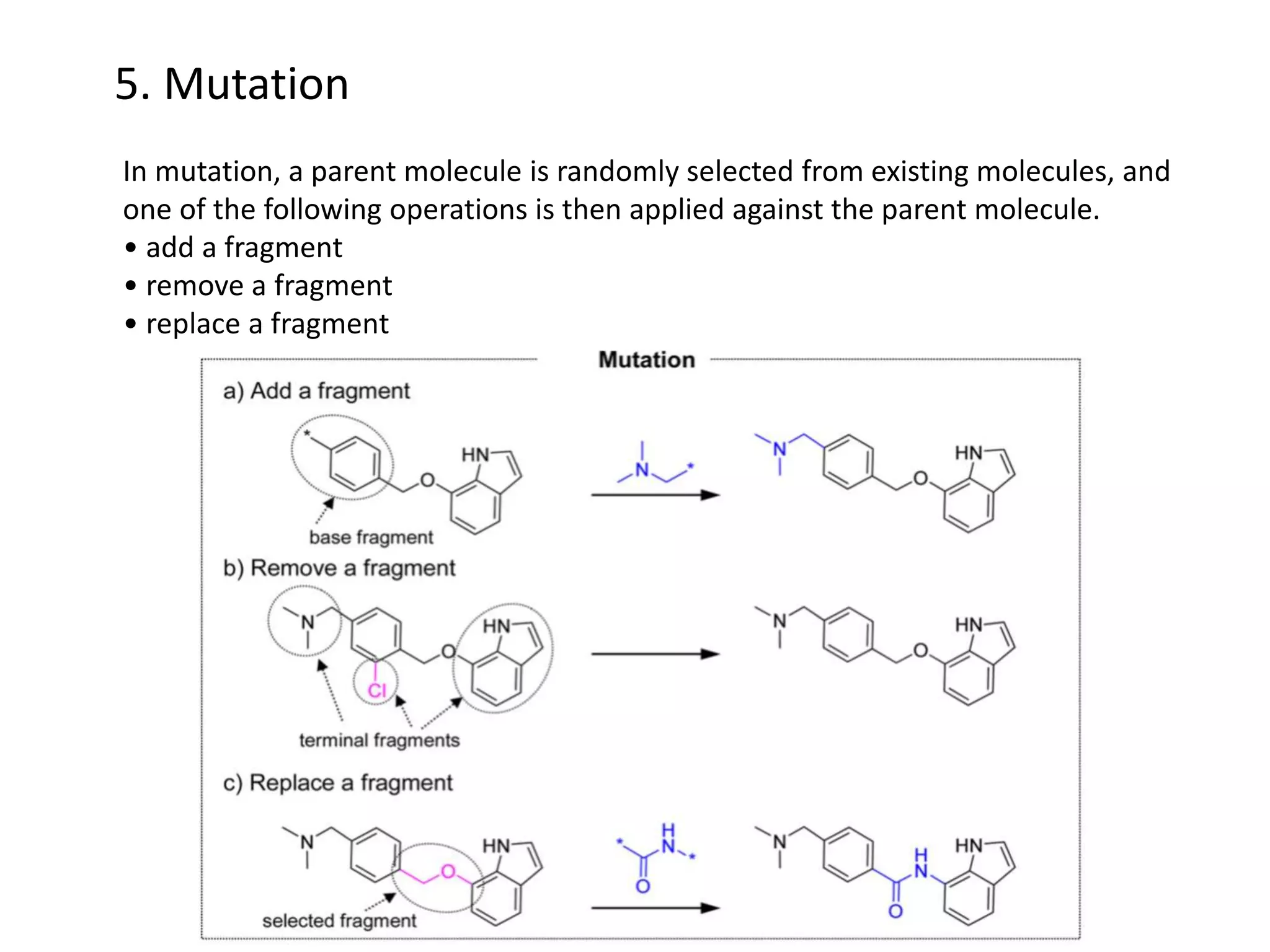
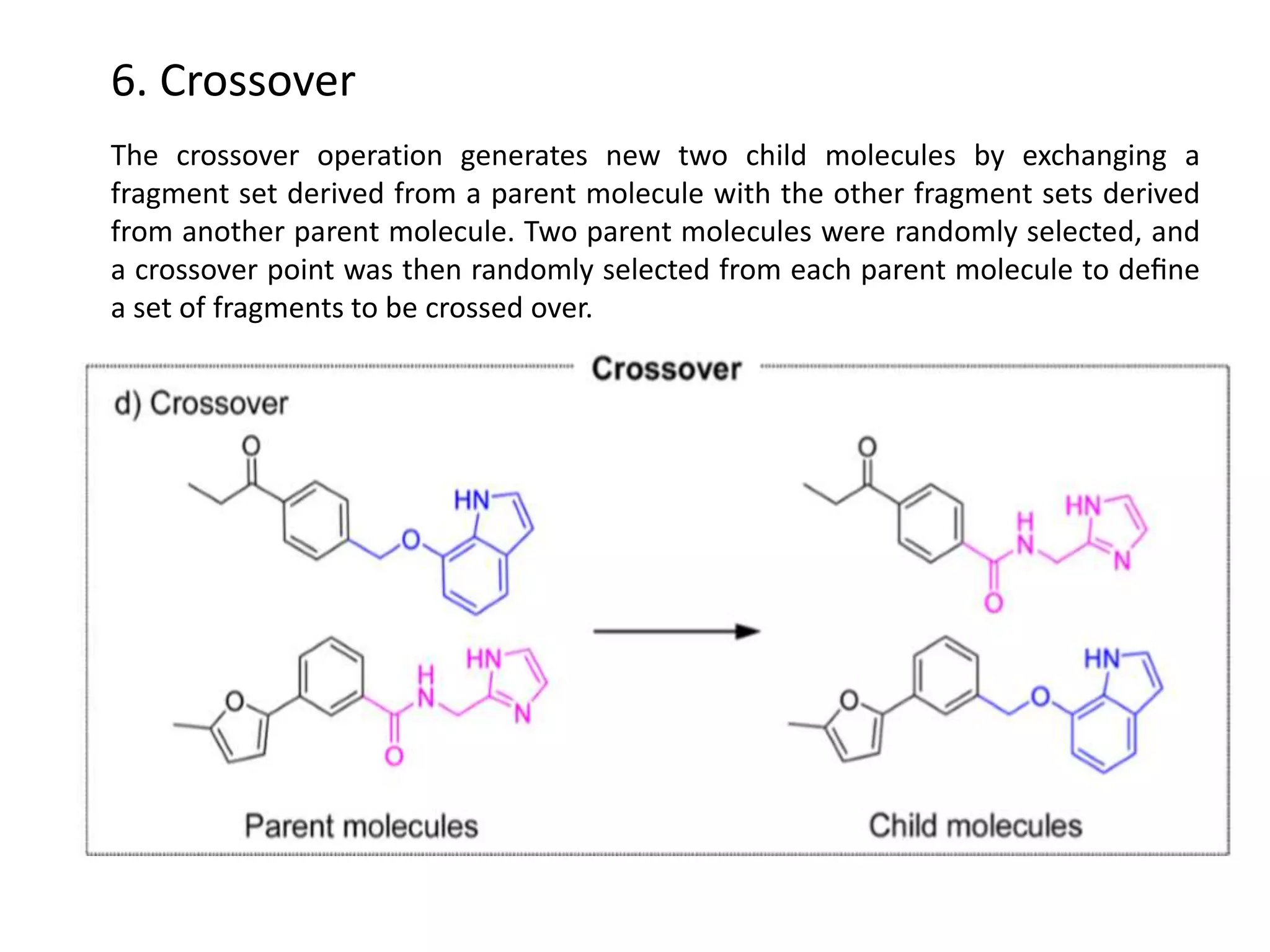
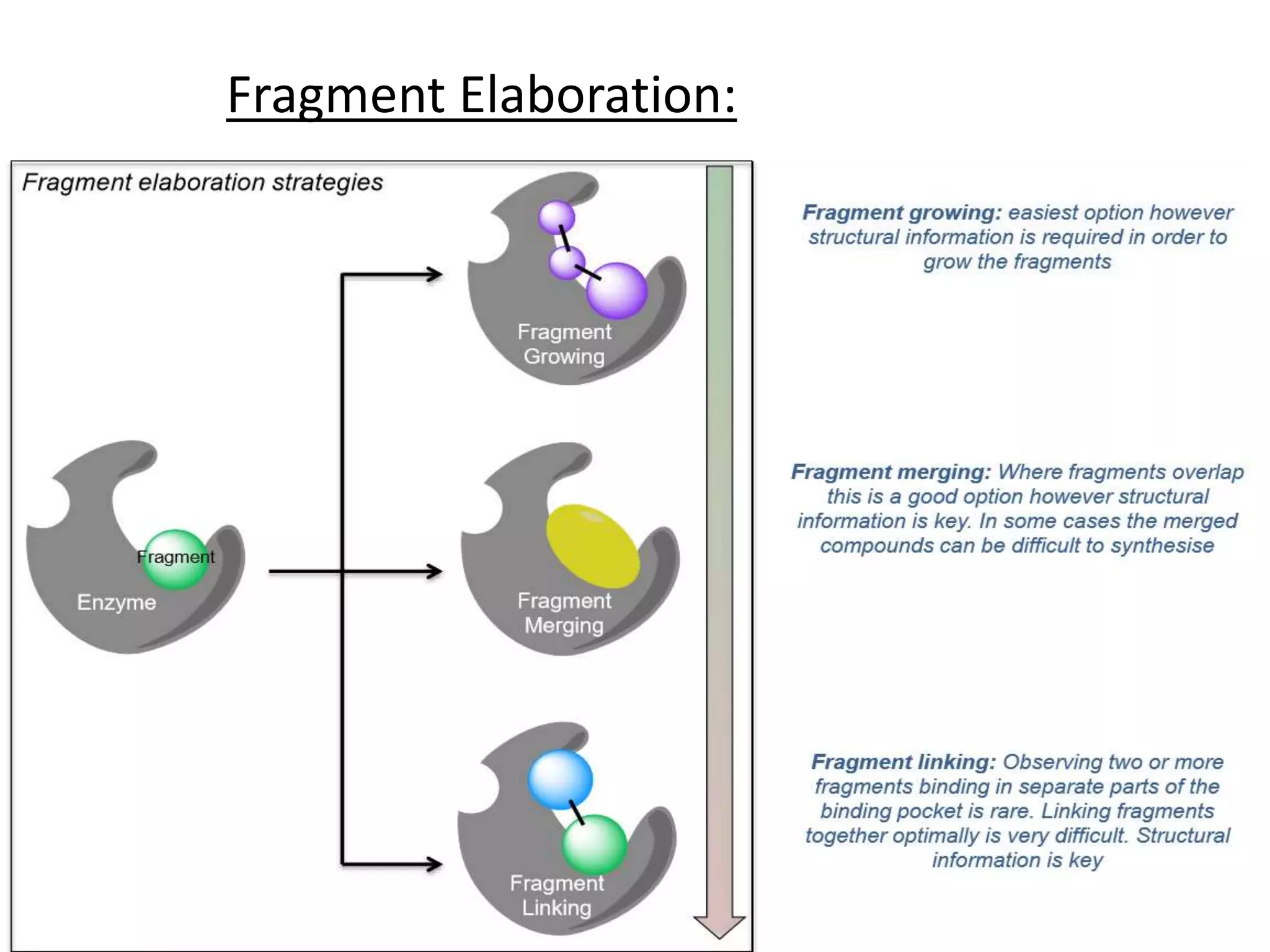
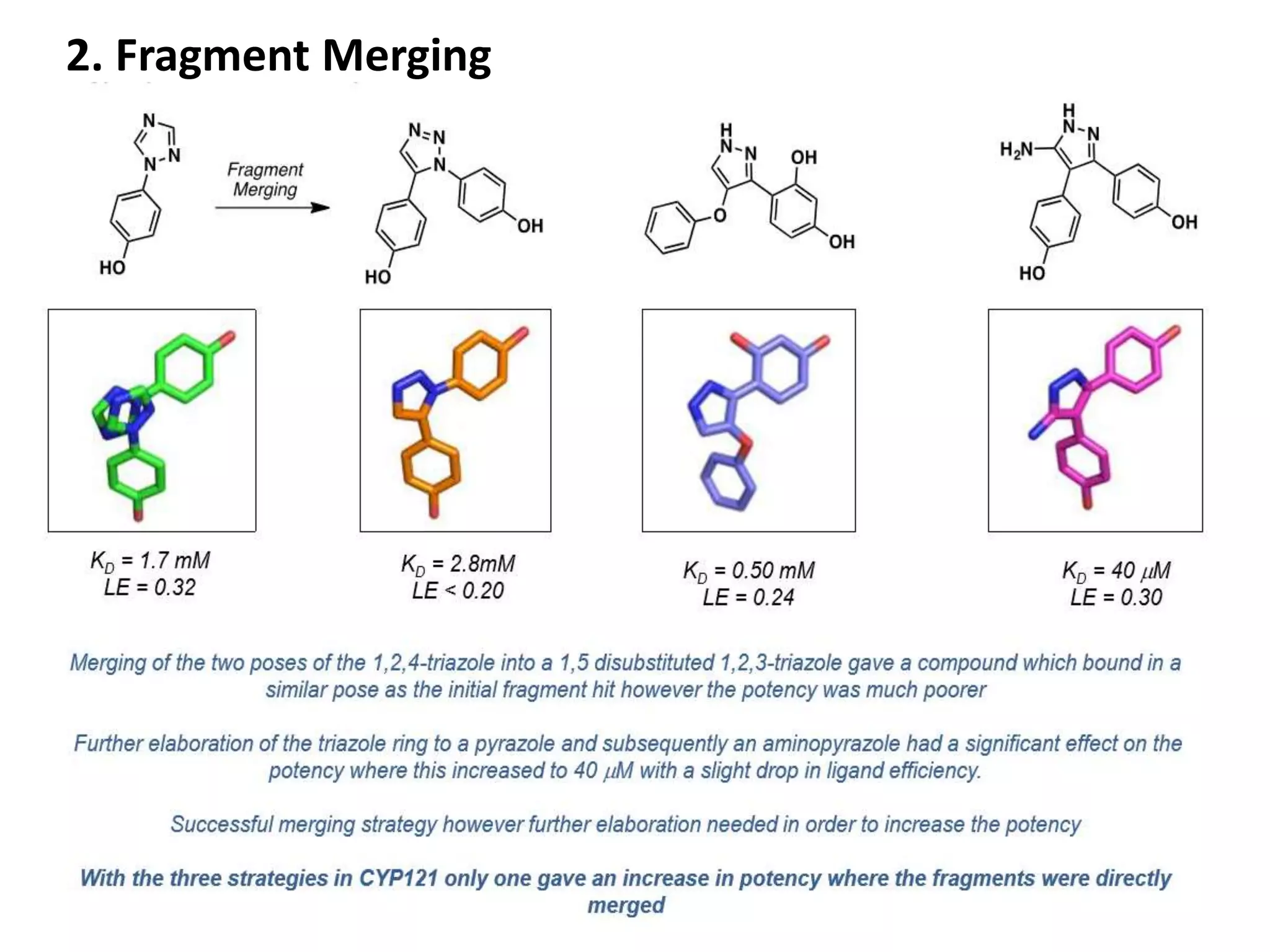
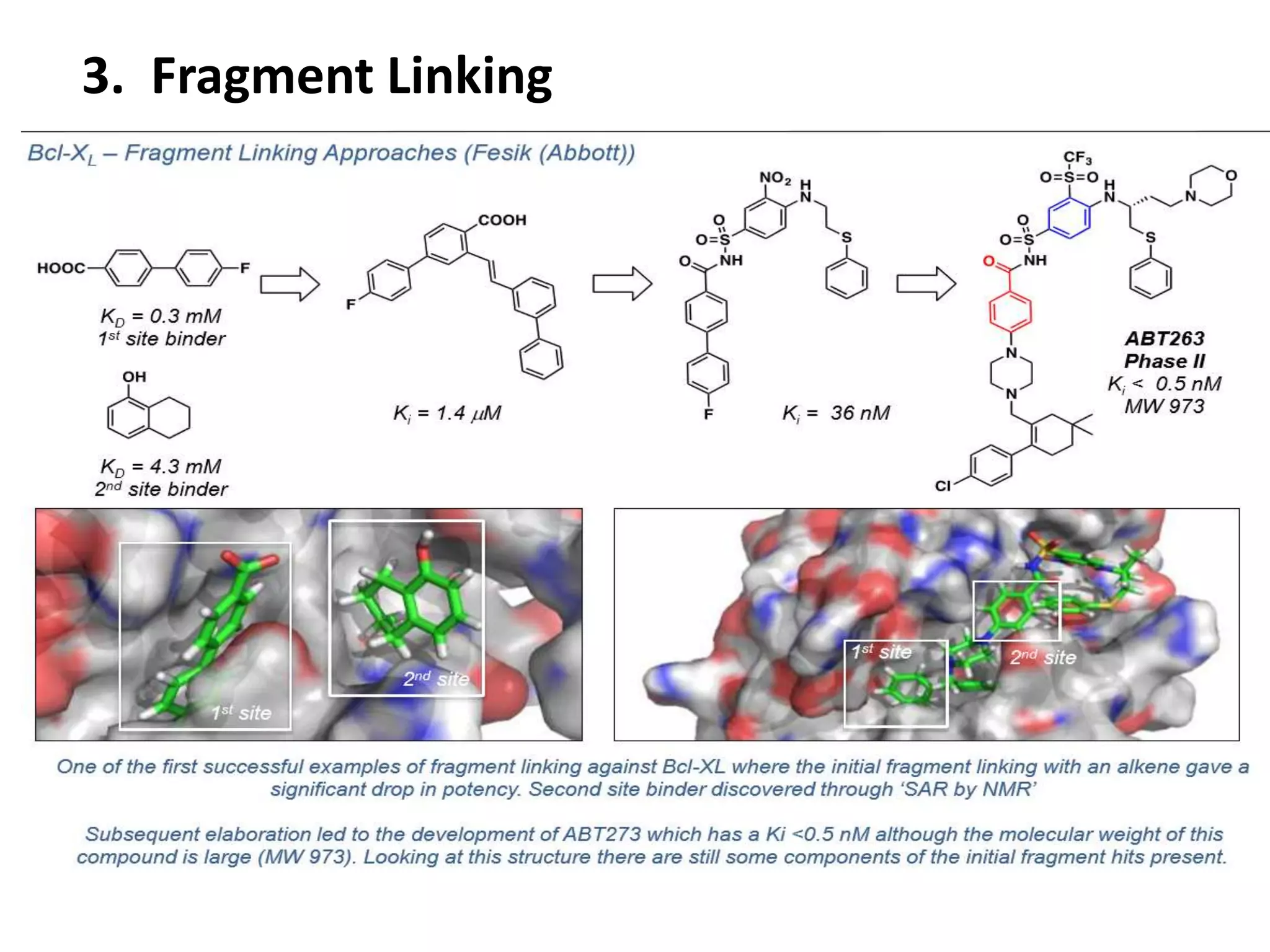
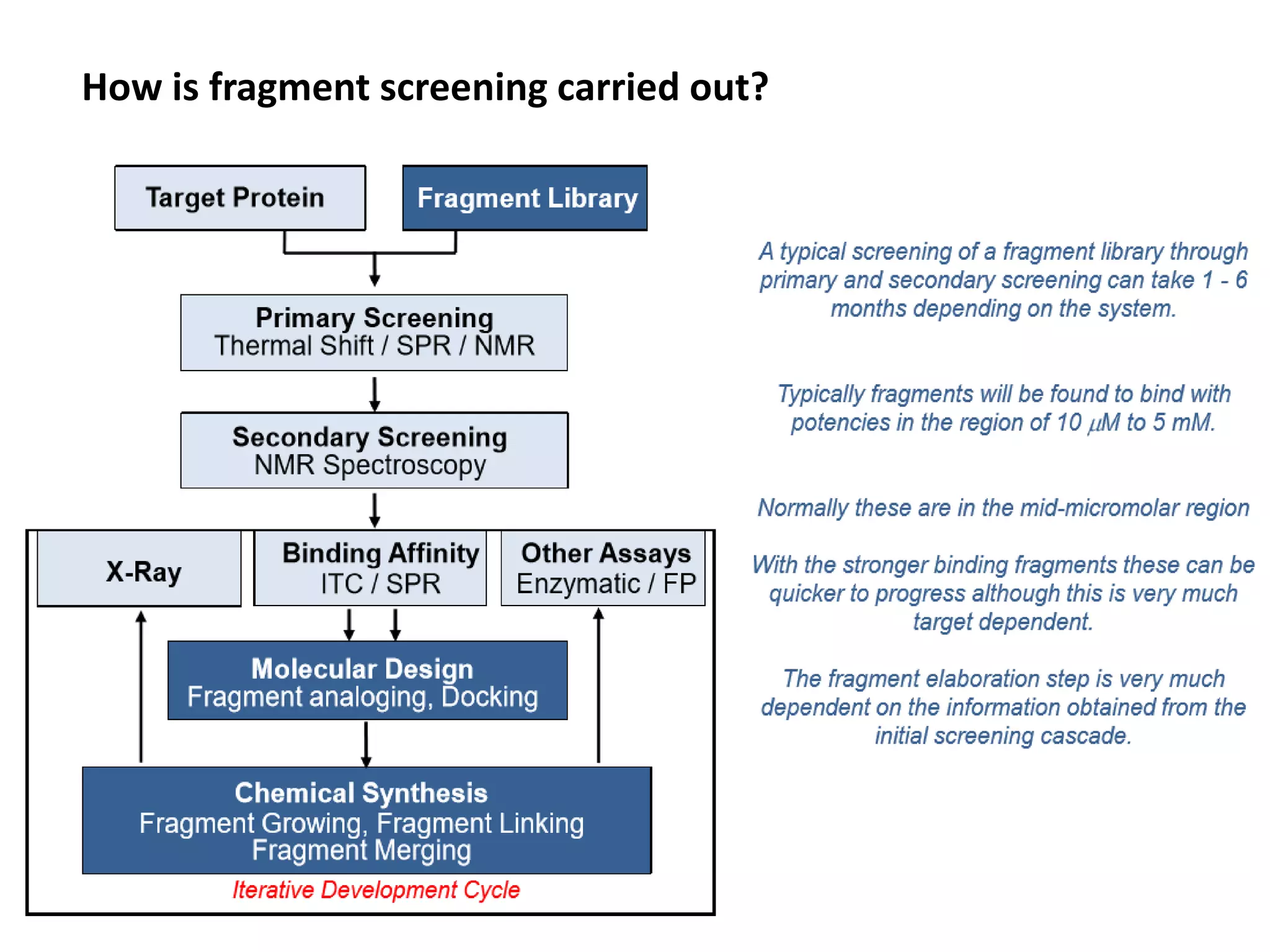
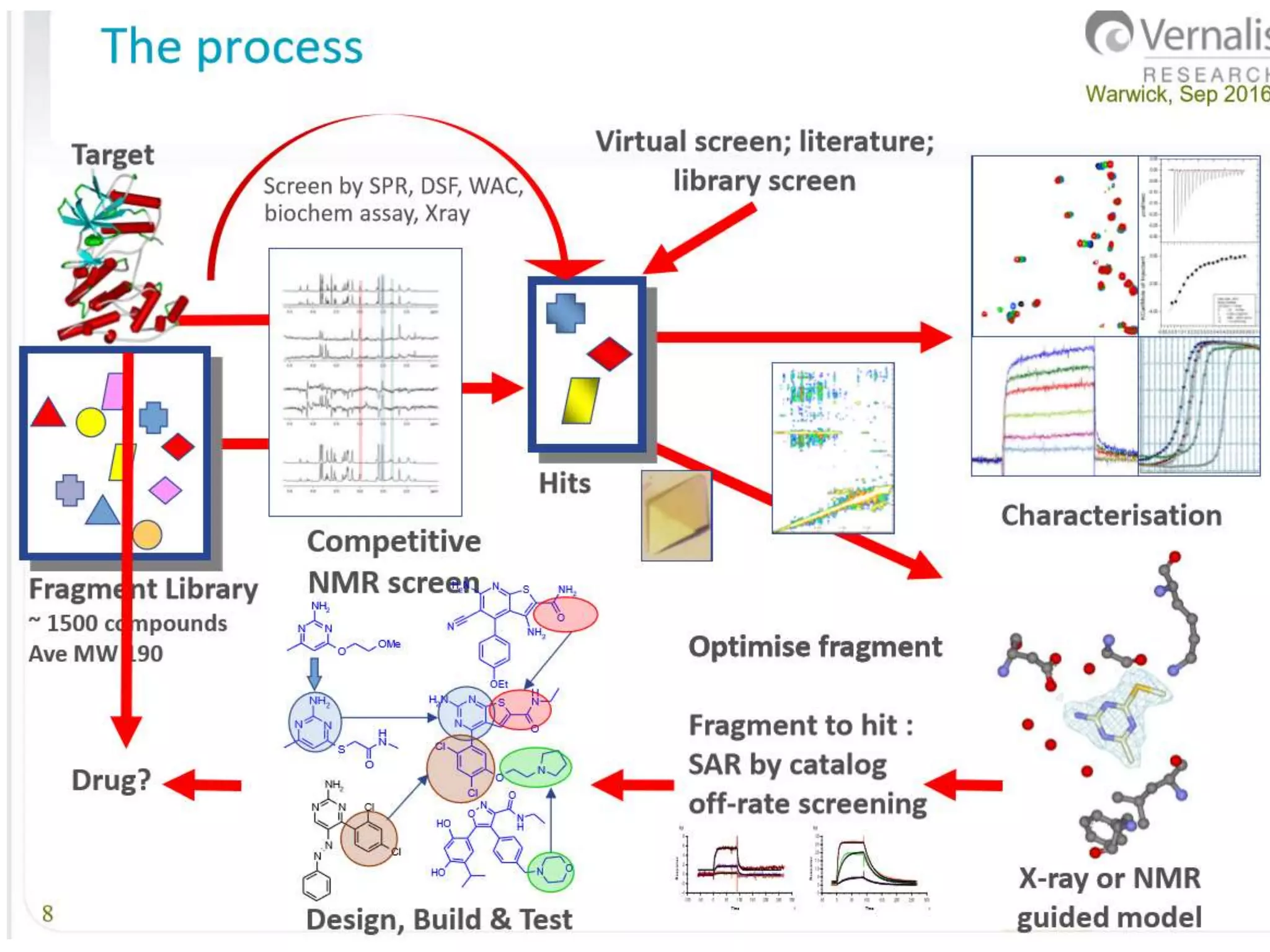
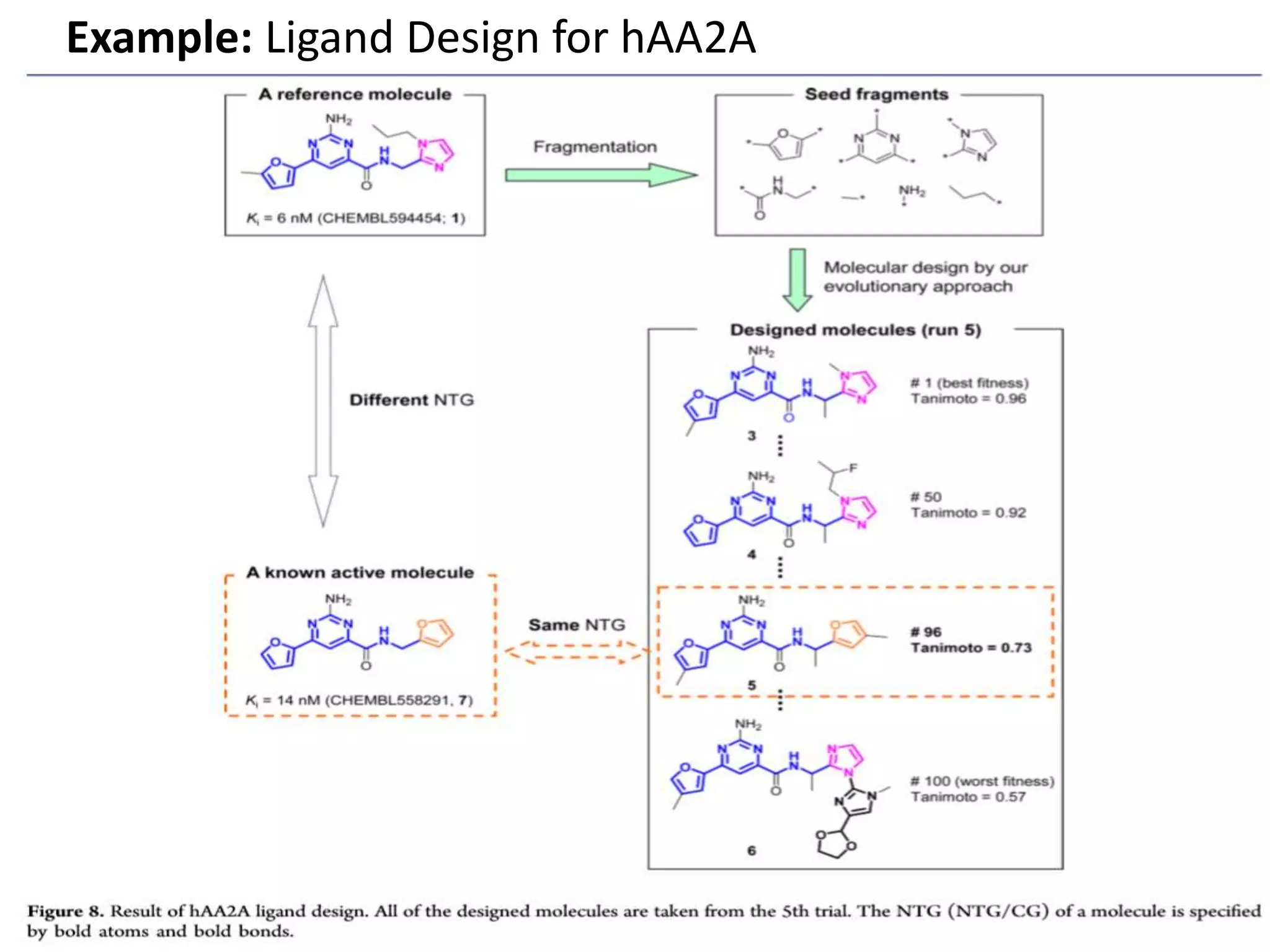
Fragment-based drug design (FBDD) uses small molecular fragments that bind weakly to a target protein's binding site. These fragments can then be grown, merged, or linked to improve binding affinity. FBDD provides starting points for challenging targets like protein-protein interactions. It increases the use of biophysics to characterize compound binding. FBDD also gives small research groups access to tools for identifying chemical probes of biological systems.





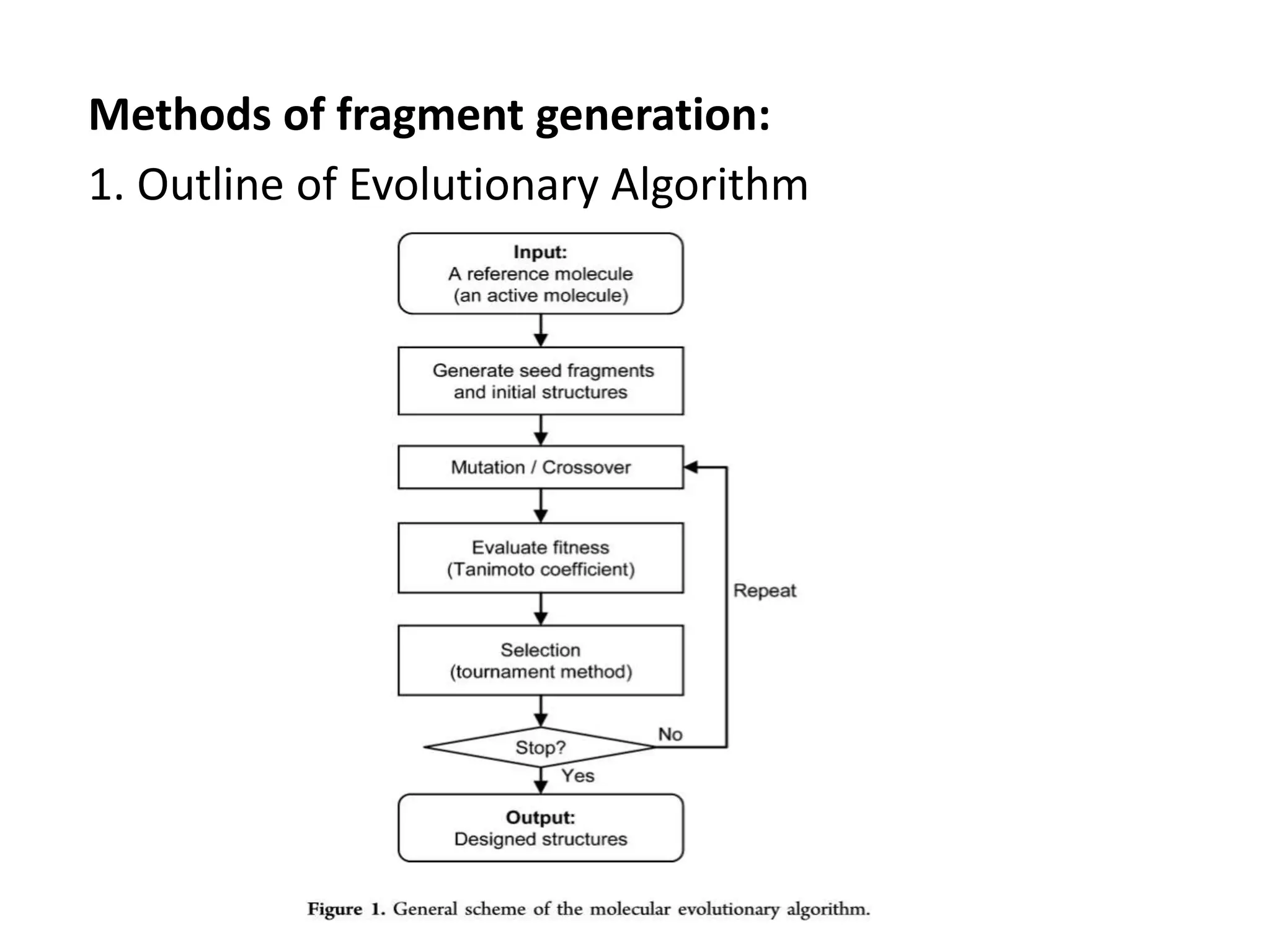
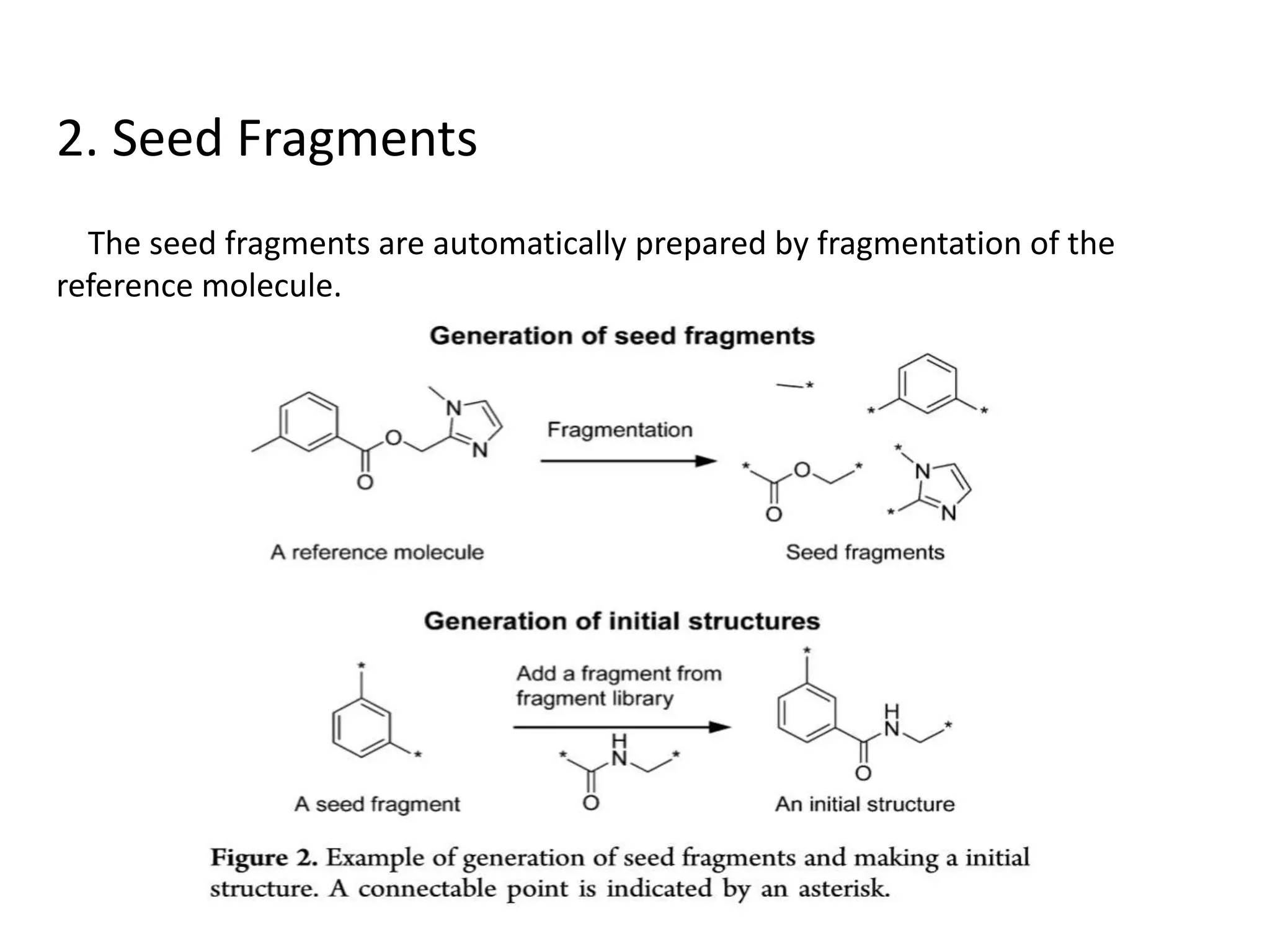
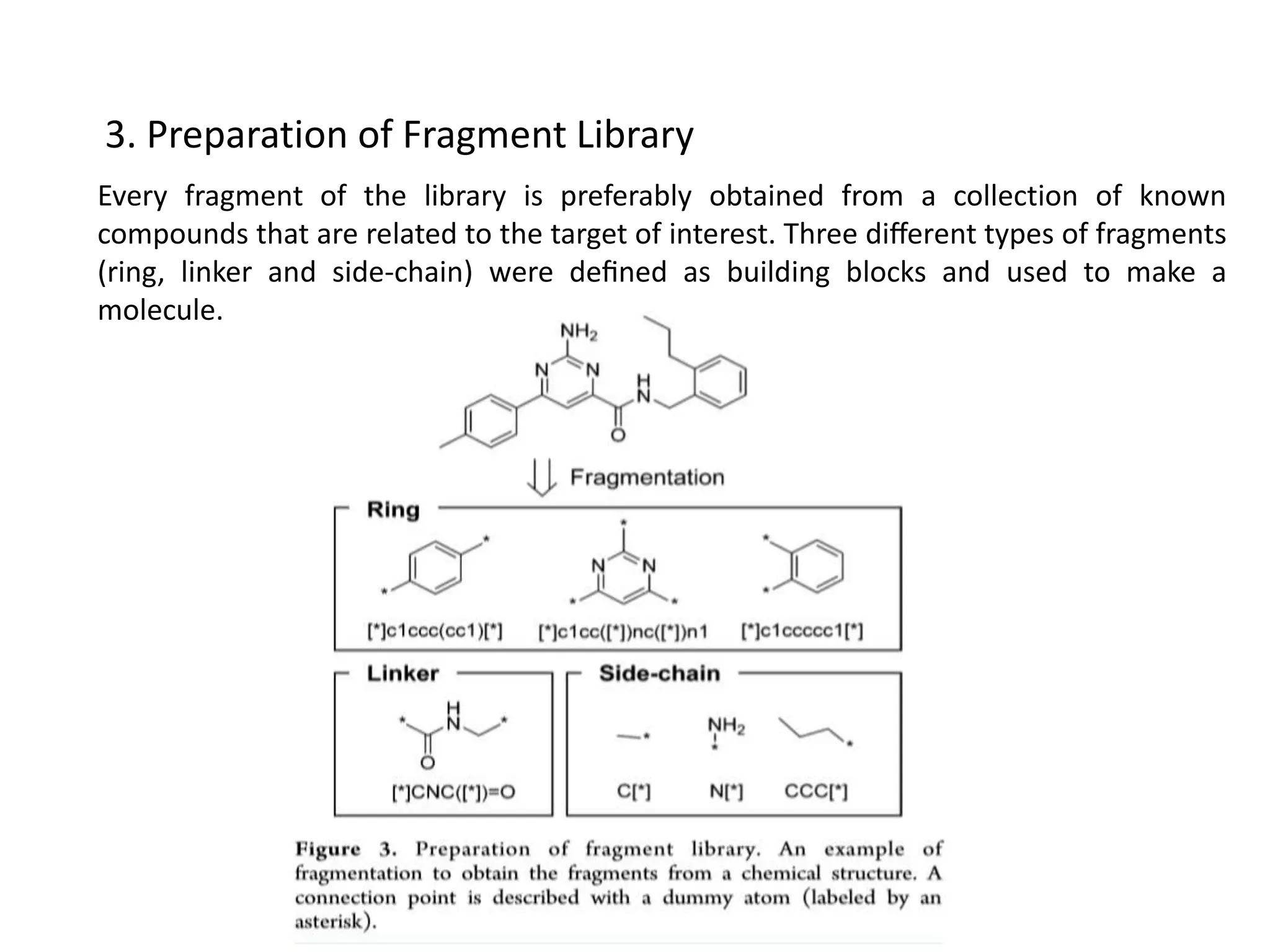
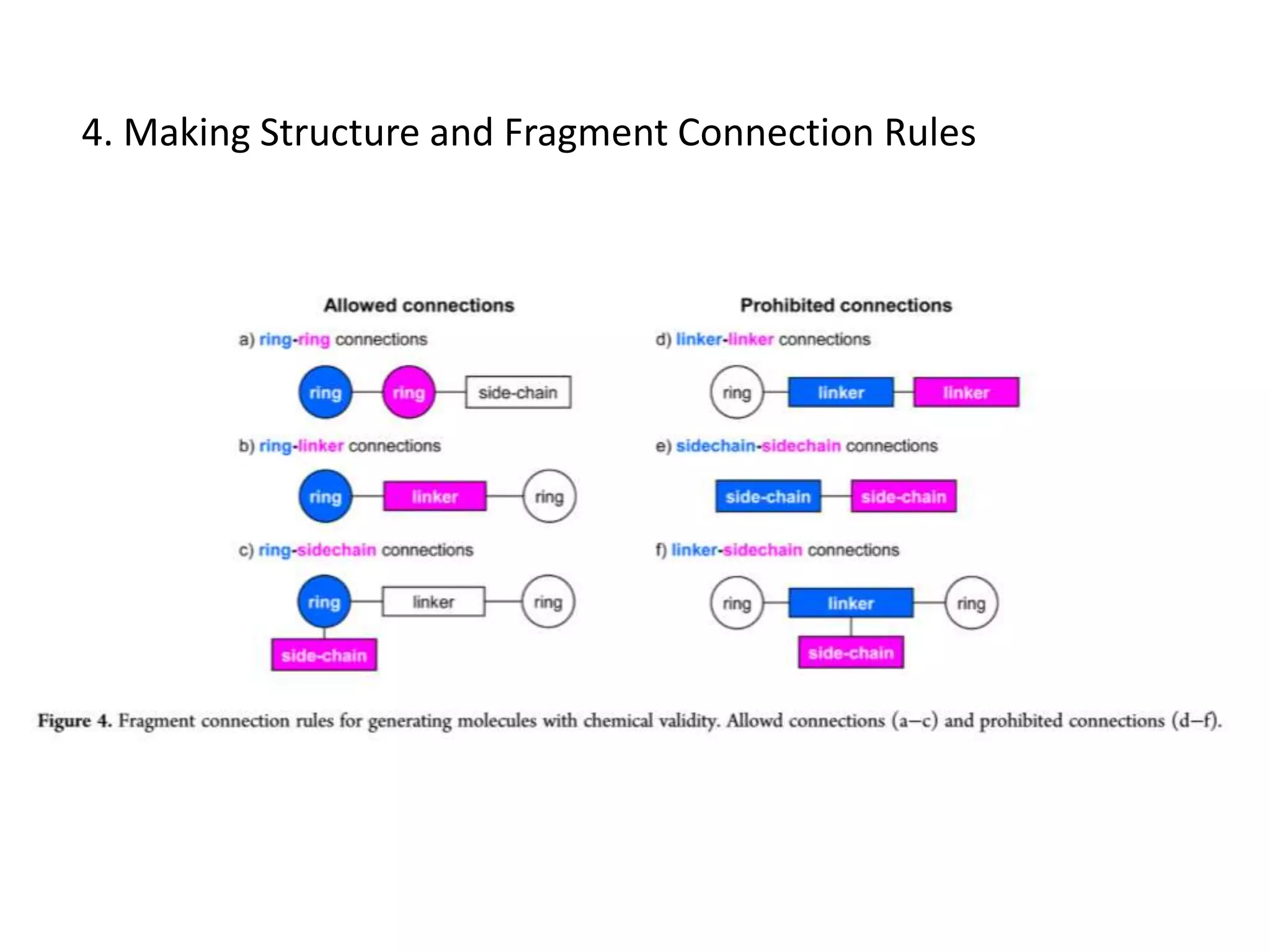






























![PEPTIDOMIMETICS [M.PHARM, BSC, MSC, B.PHARM]](https://cdn.slidesharecdn.com/ss_thumbnails/53-191219102328-thumbnail.jpg?width=640&height=640&fit=bounds)









































