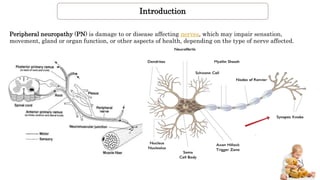
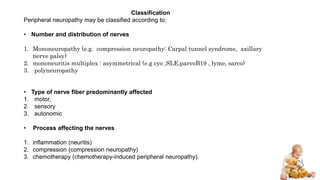
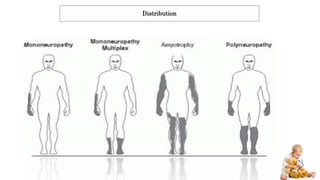
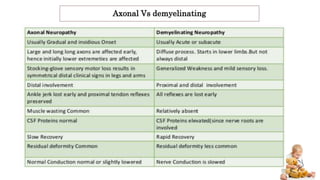
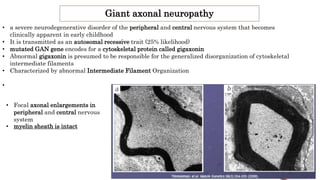
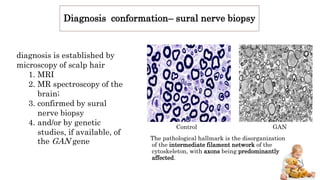
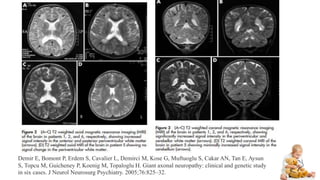
Peripheral neuropathy can be classified based on the number and distribution of affected nerves, type of nerve fiber involved, and cause. Giant axonal neuropathy is a rare genetic disorder characterized by abnormal intermediate filament organization in axons, leading to focal axonal enlargements. It presents in childhood with signs of central and peripheral nervous system involvement such as cerebellar ataxia, muscle weakness, and loss of sensation. Diagnosis involves nerve biopsy and genetic testing. Management focuses on preventing complications and optimizing development, though most patients become wheelchair-bound by their teens and deceased by their 20s.

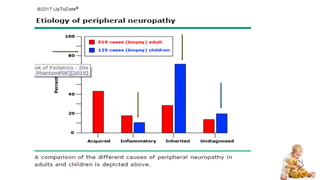
![Causes
Genetic diseases
Friedreich's ataxia, Fabry dis,CMT hereditary neuropathy with liability to pressure palsy , HMSN
Metabolic and endocrine diseases:
diabetes CRF, porphyria, amyloidosis, liver failure, hypothyroidism
Toxic & drugs:
Chemo (vincristine),Abx( metronidazole, phenytoin, nitrofurantoin, isoniazid ) heavy metals
,excess intake of vitamin B6 (pyridoxine).
Inflammatory diseases:
Guillain–Barré syndrome,[22] systemic lupus erythematosus, leprosy, multiple sclerosis,[22] Sjögren's
syndrome, Babesiosis, Lyme disease,[22]vasculitis,[22] sarcoidosis
Vitamin deficiency states: Vitamin B12 (Methylcobalamin),[22] vitamin A, vitamin E, vitamin B1 (thiamin)
Physical trauma(compression, pinching, cutting)
Others: electric shock, HIV,[22][29] malignant disease, radiation, shingles, MGUS (Monoclonal gammopathy of
undetermined significance).[30]](https://image.slidesharecdn.com/giantaxonalneuropathy-170329120223/85/Giant-axonal-neuropathy-5-320.jpg)




![Genetic counseling.
GAN is inherited in an autosomal recessive manner : At conception:
each sib of an affected individual has:
1. a 25% chance of being affected,
2. a 50% chance of being an asymptomatic carrier,
3. a 25% chance of being unaffected and not a carrier.
Carrier testing for at-risk relatives and prenatal testing for pregnancies at
increased risk are possible if the GAN pathogenic variants in a family are
known.
Preimplantation genetic diagnosis (PGD) may be an option for some families in which
the GAN pathogenic variants have been identified.
Koop O, Schirmacher A, Nelis E, Timmerman V, De Jonghe P, Ringelstein B, Rasic VM, Evrard P, Gärtner J, Claeys KG,
Appenzeller S, Rautenstrauss B, Hühne K, Ramos-Arroyo MA, Wörle H, Moilanen JS, Hammans S, Kuhlenbäumer G.
Genotype-phenotype analysis in patients with giant axonal neuropathy (GAN). Neuromuscul Disord. 2007;17:624–
30. [PubMed]](https://image.slidesharecdn.com/giantaxonalneuropathy-170329120223/85/Giant-axonal-neuropathy-15-320.jpg)





































































