This document proposes a "consensus model" approach to predict the structure of viral surface proteins using multiple sequences. It generates structural decoys for 16 diverse sequences of 5 viral proteins using Rosetta. It clusters the decoys and identifies a "consensus cluster" representing the common structure shared among the diverse sequences. Evaluation on benchmark proteins shows this approach can capture the overall protein fold despite genetic distance between sequences. This consensus structure may help design immunogens to elicit broadly neutralizing antibodies against viruses.
![New strategy for the viral protein structure predictions: “Consensus
model” approach to take advantage of sequence diversity
Introduction
The viral surface proteins are good targets for the vaccine development. They are major
targets for neutralizing antibody against viruses but elicitation of broadly reactive neutralizing antibody
(brNAb) is proved to be difficult. The structural information for these viral surface proteins is extremely
important for rational design for immunogen targeting these viruses in order to understand conserved
surface residues. Although drug targets such as viral proteases, reverse transcriptases are well
studied by crystallographic or NMR structural determinations in order to design effective drug against
these viruses, surface proteins are not as much studied as drug target proteins. Some well-studied
surface proteins such as HIV gp120 and influenza hemagglutinin show significant sequence diversity
and shown to be challenging target for elicitation of brNAb. The peptide based immunogen and
epitope scaffold-based approaches utilizing known brNAb epitope are not successful to elicit brNAb
against HIV-1 and influenza so far [1,2], and the use of minimal-sized rigid protein of conserved
sequence region in the native structural context would be necessary [3]. In order to rationally design
such immunogen, structural information for the region of protein that can fold independently is crucial.
With insufficient solved structures, we need to rely on the computational structure prediction on these
proteins. The structure prediction of viral proteins is not easy task. This class of proteins does not
share high sequence similarity with known cellular proteins [4,5,6] and also solved structures of viral
proteins are sparse compared to other protein families. Thus, it is poor target for the comparative
modeling strategy. In addition to that, the sequence diversity within the same protein is extremely high
[7,8] with their high mutation rates for virus’ immune evasion and makes it a moving target for the
structure prediction (the choice of sequence may affect outcome). Some proteins show below “twilight
zone” sequence identities [9] among same protein from different strains but same specie. In these
cases, first of all, even it is not clear what is the principle of choosing target sequence for prediction,
as even consensus sequence is just another variation. Thus, it is also true that these proteins are
difficult target for de novo prediction. In addition to extensive diversity within same protein, the
sequence space of viral proteins is vastly large yet not overlapping enough with “cellular”
(prokaryotic/archaeal/eukaryotic) sequence spaces [5,6,10]. Thus, predicting the protein structure
with knowledge-based algorithms [11,12] appears to be difficult including both comparative and
fragment assembly based free modeling strategies. As completely physics-based modeling is still not
feasible or practical at this time [13], in order to predict the structure of viral proteins, we need to use
currently available algorithms [14,15]. Although overall sequence spaces of cellular and viral proteins
may not overlap very much and unique to each other except some exchanged genes between two,
still fragment library has short “viral” sequences (such as 9mers for Rosetta fragment library) in some
degree even assembled from database composed of mostly cellular proteins. It is assumed that due
to physicochemical constraints, same sequence in both cellular and viral proteins should share similar
ensemble of structures as fragments. For this reason, physics-based fragment assembly algorithms
such as Rosetta should be able to produce decoys with same fold as native protein, probably with low
frequency.
The strategy for computational structure prediction of those viral proteins was developed using
simple principle in order to address difficulty associated with high diversity and uniqueness of viral
sequence space. Despite genetic distance, same protein must retain its function, thus same structure
(similar enough to have intact functionality). Although human papillomavirus E7 protein C-terminal
domain only shares as low as 15% sequence identity among strains (data not shown), all variants are
functional protein from isolates. Thus, in principle, they should share same fold among genetically
distant sequences, even each member of assembled sequences would generate considerably
different set of decoy population. By incorporating farthest sequence pairs among the sequences
from same protein in order to minimize overlapped structure space belonging to each sequence, this
strategy is designed to capture the common fold being populated by maximum number of sequences.](https://image.slidesharecdn.com/ede660b6-4afe-4783-9ca5-bbd0e2108ecb-141223002101-conversion-gate01/85/Viral-Protein-Structure-Predictions-Consensus-Strategy-1-320.jpg)
![It may not be abundantly populated members nor low energy structure, but common among all
sequence space and it is the “consensus model”. The multiple sequence alignment information has
been used to obtain the hydrophobic core forming residues or to improve secondary/tertiary structure
predictions and reported successful improvement of predicted structures [16,17] but the use of
different sequences within same protein directly has not been reported, appears to be mainly due to
negligible sequence diversity for cellular proteins. Utilizing Rosetta for decoy generation, 5 small viral
proteins with solved structures were used as the benchmark proteins for evaluation of this strategy.
Increase of computational power in workstation allows such “redundant” approach with manageable
time scale with affordable resources. Total of 800,000 decoys (10,000x16x5) were generated and
clustered the decoys with their pairwise TM-score [18] using just two 8-core CPU equipped
workstations. For this approach, we have chosen to use computationally expensive TM-score instead
of RMSD (this step is the most time consuming step for all process) in order to capture the overall
“fold” [19,20] rather than atomic details. In this report, first, we show the results of Rosetta generated
decoys and TM-score/energy score relations of these benchmark viral proteins then show the
performance of “consensus” approach.
Methods
Selection of benchmark proteins
The 5 small viral proteins with known structure
and variable degree of sequence diversity have been
chosen for this study to test “consensus” approach.
Availability of solved structure, sequence length below
100 residues and extremely to mildly diverse
sequences are the criteria for selection of target
proteins. We have chosen HIV p24 C-terminal
dimerization domain, vpr and vpu cytoplasmic domain,
HPV E2 DNA binding and E7 C-terminal domains that
matches above defined criteria. The 16 sequences
from each protein are chosen to be most distant in pairwise and in average. Pairwise sequence
identity among the target sequences ranges from 15 to 92% and averages pairwise identity ranges
are from 33 to 79% among same protein sequences. Table 1 illustrates sequence statistics of target
proteins. Sequences were obtained from Los Alamos HIV database (http://www.hiv.lanl.gov/)
(HIV p24/vpr/vpu) or manually collected from GenBank [21] (HPV E2/E7) and subjected to multiple
alignment by Clustal W [22] and T-Coffee [23]. Sequences are then trimmed down to the domains
with structure assessments and N- or C- terminal flexible regions in solution structure were removed
from target sequences.
Decoy generation, calculation of pairwise TM-scores and clustering
The sets of 10,000 decoys are generated for each sequence using Rosetta 2.2 without full-
atom relaxation step. Each decoy dataset was then subjected to the calculation of pairwise TM-score
of all decoy combinations. TM-scores were calculated by in-house program written in C (with TM-
align source code ported to C). Then decoys were clustered by decoy-decoy pairwise TM-scores.
Clustering was done with in-house program based on algorithm from Cluster 3.0 software source
code [24,25] by complete linkage clustering with threshold of TM-score 0.5. The clusters are further
examined for distances among all cluster members of two clusters. If all distance among members
are above threshold, they were merged. TM-scores against solved structures (structures used as
native are following; p24:2JYL, vpr:1M8L, vpu:1VPU, E2:1A7G, E7:2B9D) were calculated for all
decoys (1VPU has 9 models in PDB file so that all 9 models were used for the calculation) and
Rosetta energy scores were extracted From Rosetta silent files. All calculations were executed on 2
of 2.66GHz dual quad-core Xeon equipped Mac Pro with Mac OSX 10.6 and all programs were
compiled with Intel compiler v11.1 with full optimization flags.
Calculation of consensus cluster
Table 1
Sequence ID (%)Length
mean Max Min
P24 70 79.3 92 62
vpr 56 72.4 89 50
vpu 81 57.7 82 41
E2 84 40.1 90 22
E7 44-54 33.3 64 15](https://image.slidesharecdn.com/ede660b6-4afe-4783-9ca5-bbd0e2108ecb-141223002101-conversion-gate01/85/Viral-Protein-Structure-Predictions-Consensus-Strategy-2-320.jpg)

![which is rough threshold for the same fold [20]. Thus it is difficult to select the decoy with TM-score
beyond 0.5 (against native
structure) for final model.
Clustering of decoys
The clustering of decoys for
these proteins are performed with
hierarchical complete linkage
clustering algorithm since each
cluster should only contain
structurally close enough (same
fold) decoys in order for capturing
consensus cluster later.
Therefore, TM-score 0.5 was set
as threshold for complete linkage
clustering. The number of clusters
varies to a great extent among the
proteins and sequences. HPV E2
has significantly more clusters
(table2) with threshold value 0.5
(mean 276.5) in comparison with
HIV vpr (mean 12.5). Number of
clusters of HPV E7 fluctuates greatly (31-432) by sequence in same way as energy/TM-score plots.
Similar trend is also observed by coverage of Top 10 clusters (Sum of top 10 cluster members
divided by all decoys). Threshold value 0.3 for the clustering Top 10 decoys were determined as the
value giving enough cluster members for choosing “consensus” cluster but not to include too diverse
structural ensemble. The threshold value of 0.5 (used for initial clustering) produced too many
clusters with small number of cluster members among Top 10 decoy set and cannot reach
“consensus” as each cluster has too incomplete coverage of sequences. In order to determine
consensus cluster, TM-score threshold was lowered to yield enough cluster members populated by
most of sequences, but pairwise
distance among the Top 10 decoy set
and average distance from each cluster
center is not far below from 0.5. The
threshold value of TM-score 0.3 appears
to be low but clustering is performed with
complete linkage clustering algorithm so
that even the most distant decoys have
TM-score around or above 0.3. For all 5
viral proteins, the distances are within
the range of same fold as average
pairwise TM-score is 0.49 (0.46-0.51)
and 0.55 (0.51-0.57) for average
distance from cluster center for all Top
10 decoy sets. Thus, it is concluded that
these clusters are representative of
decoys in same fold and clusters with
highest sequence coverage were taken
as consensus cluster and center as
consensus model.
Consensus model strategy
Table 2
p24 vpr vpu E2 E7
Seq# Clust cover. Clust cover. Clust cover. Clust cover. Clust cover.
1 65 0.816 9 0.999 97 0.701 224 0.480 432 0.165
2 62 0.800 8 0.999 60 0.803 411 0.372 54 0.893
3 41 0.802 12 0.997 74 0.810 292 0.322 226 0.610
4 50 0.657 67 0.809 93 0.770 320 0.380 57 0.866
5 30 0.924 8 0.999 164 0.495 145 0.700 46 0.921
6 44 0.866 10 0.999 150 0.533 303 0.323 113 0.767
7 32 0.868 8 0.999 103 0.671 345 0.295 56 0.916
8 51 0.584 12 0.994 135 0.547 237 0.614 43 0.923
9 38 0.843 8 0.999 112 0.663 293 0.360 31 0.927
10 43 0.831 6 0.999 123 0.654 267 0.398 318 0.391
11 147 0.307 19 0.976 110 0.702 235 0.503 30 0.956
12 48 0.836 7 0.999 18 0.987 368 0.469 291 0.339
13 47 0.719 8 0.999 77 0.804 255 0.605 64 0.896
14 29 0.945 6 0.999 161 0.456 257 0.514 37 0.969
15 23 0.965 9 0.999 94 0.693 227 0.720 399 0.349
16 29 0.902 3 0.998 63 0.917 245 0.455 438 0.242
Ave 48.7 0.792 12.5 0.985 102.1 0.700 276.5 0.469 164.7 0.696
S.D. 28.8 0.164 14.9 0.047 39.4 0.147 64.3 0.133 157.9 0.294
Figure 2: Aligned predicted models (brown) and solved structures
(white). The consensus models of 5 viral proteins are aligned to
their solved structures by TMalign. In the figure, “Aligned” indicates
the residues within 5 angstrom distances from solved structures.
The solved HIV vpu structure is NMR solution structure with 9
models. Displayed in the figure is model 1 which was best aligned
with consensus model. Other 4 structures are crystal structures.](https://image.slidesharecdn.com/ede660b6-4afe-4783-9ca5-bbd0e2108ecb-141223002101-conversion-gate01/85/Viral-Protein-Structure-Predictions-Consensus-Strategy-4-320.jpg)

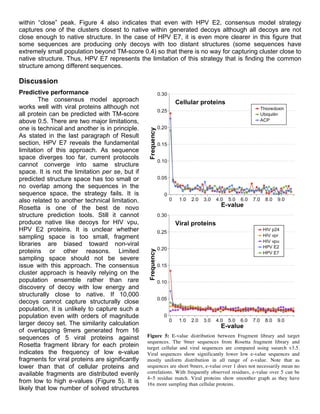
![of viral proteins compared with other protein classes results in small population of fragments with low
e-value. Although it is not necessarily true that availability of low e-value fragments guarantees
accurate predictions, opposite would be true. The distant fragment library sequences from actual
protein are likely to have lower structural correlation to actual structures. These libraries had to be
used for fragment assembly and likely to keep decoy structures away from solved structures. As viral
proteins sequence space is generally unique compared to other protein classes, the low number of
available structures for fragment library generation would be to be blamed for the poor performance in
decoy generation of viral proteins. Put together, available sampling space is limited due to the lack of
available solved structures corresponding to the viral sequence space. Another factor related to
uniqueness of sequence can affect the secondary structure prediction used for Rosetta decoy
generation. Secondary structure of HIV vpu was not accurately predicted by all three methods
supported by Rosetta (PSIPRED, JUFO and SAM) and resultant model has extended C-terminus
rather than actual α-helical structure. Considering uniqueness of viral sequence and structure spaces,
it is not surprising that all three secondary structure prediction algorithms do not work for some viral
proteins. If secondary structure has been accurately predicted, it is likely that the result for HIV vpu
was much more accurate and well over TM-score 0.5.
Further improving performance by combining “consensus” approach with sampling of low
Rosetta energy decoys was attempted but unsuccessful. There are 2 cases (E2 and E7) which
improvement was observed by filtering the decoys by energy score but other 3 cases (p24, vpr and
vpu), filtering actually worsened the results (Figure S2). Rosetta generated the population of decoys
for HPV proteins with lower energy that is less distant to native than overall ensemble. Unfortunately,
opposite is true for HIV proteins and filtering by energy score actually worsened results for these
proteins. It is puzzling and not known why Rosetta energy and TM-scores are uncorrelated for viral
proteins. Nonetheless, due to the fact that viral proteins do not have lowest energy states with their
native structure or Rosetta energy function does not represent native viral protein energy, an attempt
to further improve results by taking low energy decoys failed. Although full-atom relaxation step was
not performed, it is unlikely that the Rosetta energy scores dramatically change with full-atom
relaxation. The largest cluster center strategy that is taken frequently also did not work as good as
consensus strategy. The largest cluster does not necessarily represents the decoy population with
smallest distance form native structure (7,6,5,1,2 cases out of 16 for p24, vpr, vpu, E2 and E7,
respectively). It is, therefore, difficult to determine the correct fold by selecting center of largest cluster
too. And again, there is problem of choosing from many different sequence variants. On the other
hand, consensus strategy took advantage of sequence diversity (ranging from 33 to 79% in mean
sequence identities) and successfully captured the cluster with most structurally close to native fold
even the case of overall decoy population is not very close to native (figure 4). An initial assumption
that short viral sequence fragments should have enough overlap with cellular protein sequence space
appears not standing and it is probable that even 9mer fragments are unique for viral sequences
compared with cellular sequences, as indicated by significantly lower number of low e-value and
abundant high e-value fragments are observed in fragment library (Figure 5).
Usefulness
This approach is developed mainly targeting the structure prediction of small viral proteins for
antigen/immunogen design. This strategy captures same or close fold (around or above TM-score
0.45) for 4 out of 5 proteins. The purpose wise, this result is a success in the use of the predicted
model for the evaluation of properties of designed antigen/immunogen such as mapping of epitope
and immunodominant region. For these purposes, atomic level of accuracy is desirable but not
necessary. In fact, this approach is used for predicting the structure of small protein based
immunogen for HIV-1 vaccine and generating quite useful information to interpret immunological and
biochemical data obtained by antigenic and immunogenic studies. Many biochemical data can be
interpreted with low-resolution structure model (correct fold) instead of high-resolution atomic models
for the application such as function or protein family inference [26]. Thus, this approach can produce](https://image.slidesharecdn.com/ede660b6-4afe-4783-9ca5-bbd0e2108ecb-141223002101-conversion-gate01/85/Viral-Protein-Structure-Predictions-Consensus-Strategy-7-320.jpg)