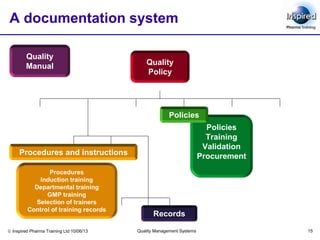
This document discusses documentation and record keeping requirements for quality management systems. It covers the key principles of documentation including accuracy, integrity and availability. It describes the types of documents and records required such as specifications, manufacturing instructions, certificates and reports. The document provides details on EU GMP chapter 4 requirements for documentation practices, document control, record retention and approval. It emphasizes that a documentation system is essential to meet regulatory standards and continual improvement.






















































![supriya.k_ppt[1].pptx documentation for QA and QC](https://cdn.slidesharecdn.com/ss_thumbnails/supriya-250809112814-eb4e6f1f-thumbnail.jpg?width=640&height=640&fit=bounds)



























