Downloaded 46 times







![ More recently, Maas et al.
proposed a modified version of the
Marshall score (termed the
Rotterdam score) to account for
additional radiographic criteria
that more accurately predict
survival from head injury.
These systems describe a
strong correlation between
CT scan findings (e.g.,
compression of the basal
cisterns, presence of
subarachnoid hemorrhage
[SAH], midline shift) and
clinical course, mortality, and
functional outcome after TBI.
• Because determining an accurate post-resuscitation GCS score is extremely
difficult in the current era, it is likely that radiographic scoring systems will play a
larger role in predicting outcome and directing care in the acute period after
TBI.](https://image.slidesharecdn.com/tbifinal-220829152721-4ec727c3/85/Traumatic-brain-injury-pptx-12-320.jpg)






























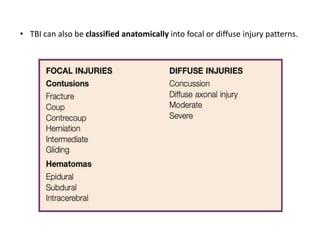
The document summarizes traumatic brain injury (TBI), including the primary mechanical injury and subsequent secondary injuries. It describes the pathophysiology of contusions, hematomas (epidural, subdural, intracerebral), diffuse axonal injury, and concussions. It discusses classification systems for TBI severity and outlines the mechanisms of injury, including contact forces, inertial loading, rotational acceleration, and angular acceleration. Factors affecting injury extent and outcomes are also summarized.

















![Management of acute ischemic stroke including tia [autosaved]](https://cdn.slidesharecdn.com/ss_thumbnails/managementofacuteischemicstrokeincludingtiaautosaved-180808183403-thumbnail.jpg?width=640&height=640&fit=bounds)


























































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)










