Downloaded 15 times





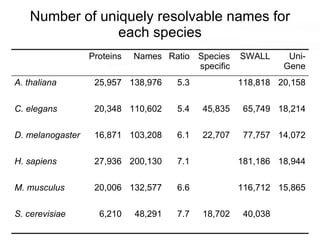



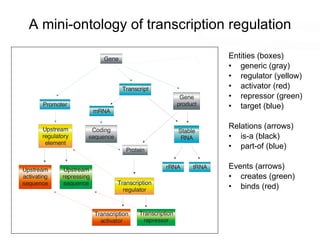


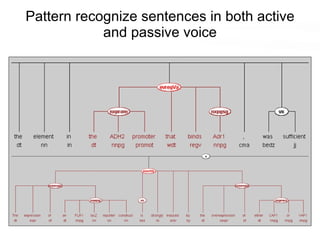






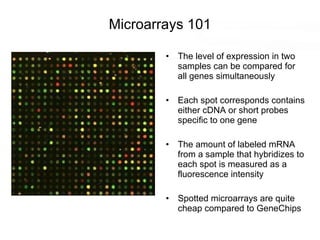
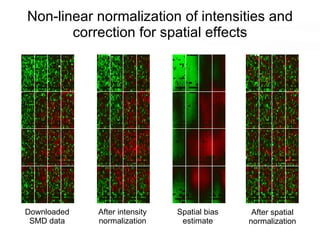


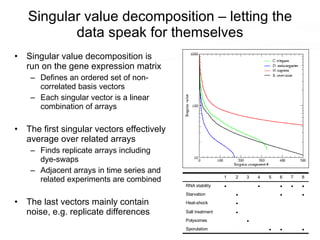
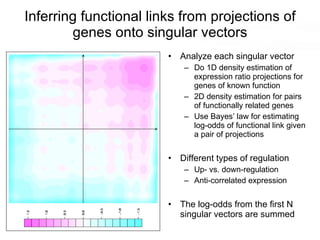





This document discusses various topics related to transcriptomics and lexico-syntactic analysis including: - The challenges of resolving gene names and identifiers from multiple databases. - Using text mining of biomedical abstracts to identify regulatory relationships between genes/proteins by parsing sentences and identifying relationships. - Integrating gene expression data from multiple experiments and species to infer functional links between genes based on correlations in expression profiles. - Other related resources discussed include the STRING database for predicting protein-protein interactions and the use of gene synonyms to integrate different types of biological data.
























![melissa Poster SGM mel[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cbc72576-c985-4bc6-9f04-631b12a7f357-160813003809-thumbnail.jpg?width=640&height=640&fit=bounds)


















































































