Download to read offline







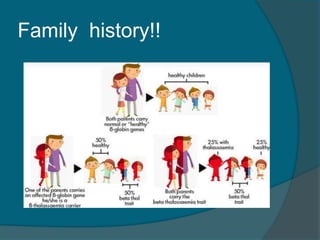






















This document discusses a case study of beta thalassemia major in a 4-year-old male child presenting with growth retardation, irritability, and anorexia from infancy. On examination, the child had pallor, hepatosplenomegaly, and no lymphadenopathy. The document then summarizes the classification, clinical features, complications, and management of different types of thalassemia including beta thalassemia major, beta thalassemia minor, thalassemia intermedia, alpha thalassemia trait, HbH disease, and hydrops fetalis. Key points covered are genetic causes, presentation from infancy, anemia, organomegaly, bone changes,










































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)












































