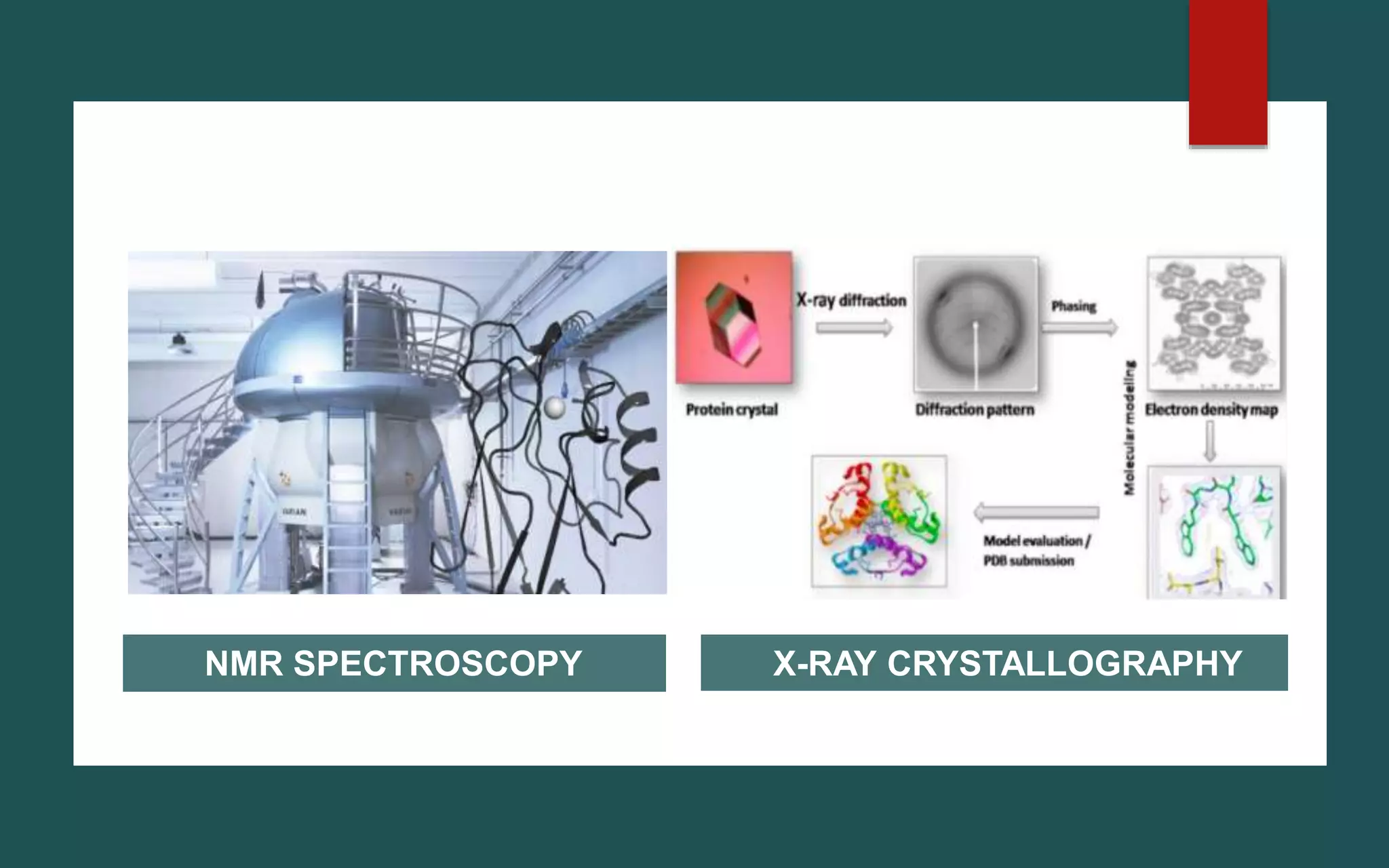
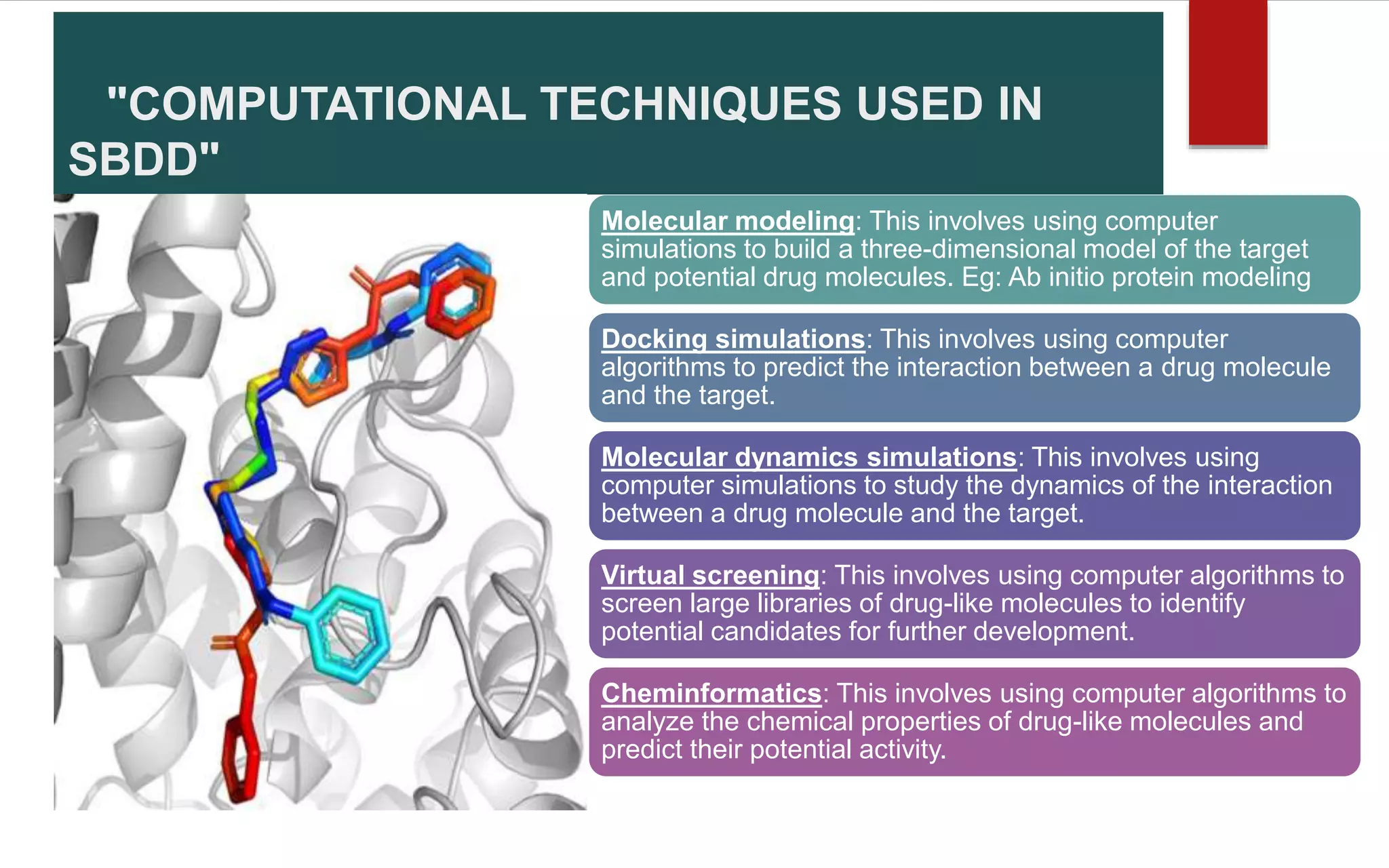
Structure-based drug design (SBDD) is a computational approach that uses the 3D structure of target proteins to guide the design of potential drug molecules. SBDD leverages knowledge of molecular interactions between drugs and proteins to design drugs more likely to bind to targets and exert therapeutic effects. Computational techniques like molecular docking, dynamics simulations, and virtual screening are used to model interactions and screen large libraries of compounds. Laboratory methods like X-ray crystallography and NMR spectroscopy provide protein structures to inform computational modeling. SBDD has potential to increase drug discovery efficiency and success rates by enabling rational drug design focused on target binding and properties.