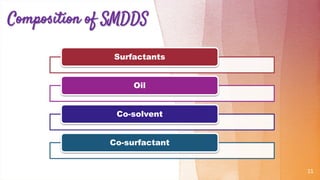
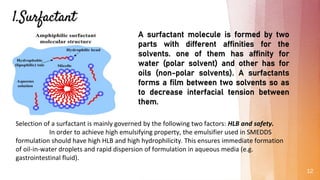
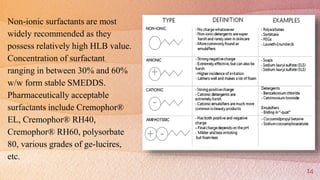
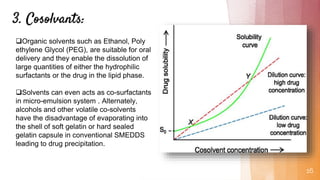
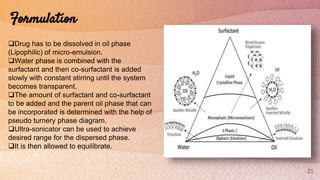
Self-microemulsifying drug delivery systems (SMEDDS) are isotropic mixtures of natural or synthetic oils, solid or liquid surfactants, and hydrophilic solvents/co-surfactants that form fine oil-in-water microemulsions upon mild agitation followed by dilution in aqueous fluids such as gastrointestinal fluids. The main purpose of SMEDDS is to improve the oral bioavailability of poorly water-soluble drugs. Key components include oils, surfactants such as Cremophor EL and polysorbate 80, and cosolvents/co-surfactants such as PEG 400 and ethanol. SMEDDS formulations are evaluated using methods such as visual assessment, differential scanning cal






















![evaluation
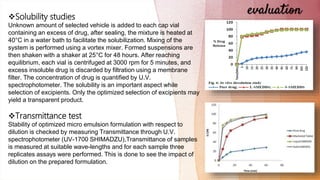
Stability:
1. Temperature Stability
Shelf life is a function of time and storage temperature is evaluated by
visual inspection of the SMEDDS system at different time period.
SMEDDS is diluted with purified water and to check the temperature
stability of samples, they are kept at three different temperature range
[2-8°C (refrigerator), room temperature etc.] and observe for any
evidence of phase separation, flocculation or precipitation so that an
appropriate storage condition may be prescribed for the developed
product.
2. Centrifugation
In order to estimate meta-stable system, the optimized SMEDDS
formulation is diluted with purified distilled water. Then micro emulsion
is centrifuged at 1000 rpm for 15 minute at 0°C and observed for any
change in homogeneity of micro emulsions. This indicates the stability
of product upon dilution.](https://image.slidesharecdn.com/smeddsarifseminar-201213095211-230303123644-c8108c55/85/smeddsarifseminar-201213095211-pdf-28-320.jpg)



































![SELF MICRO EMULSIFYING DRUG DELIVERY SYSTEM [SMEDDS]](https://cdn.slidesharecdn.com/ss_thumbnails/sagarsavalepaper-150914074813-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)





































