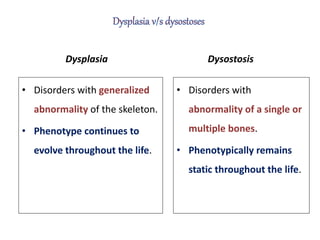
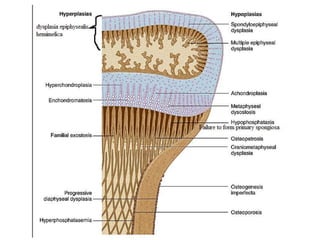
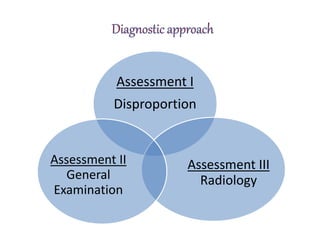
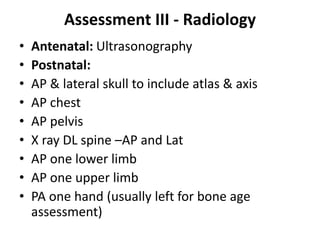
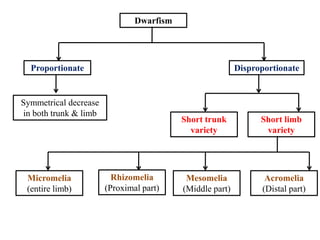
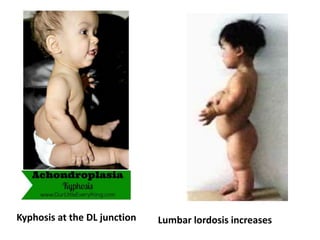
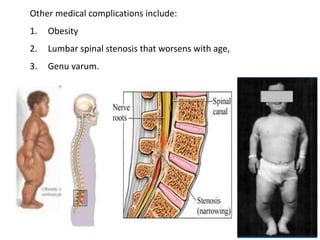
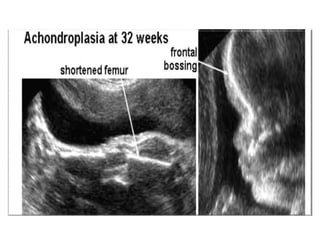
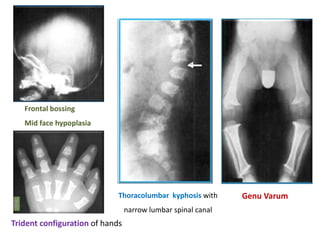
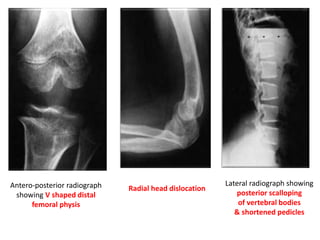
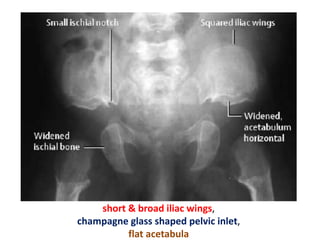
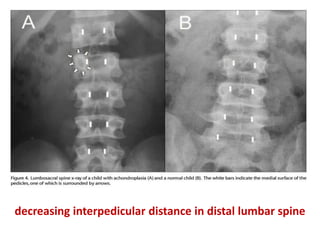
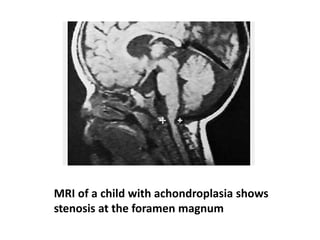
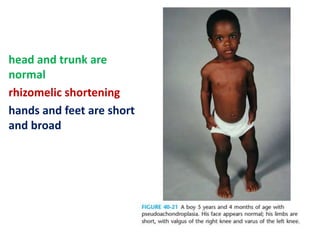
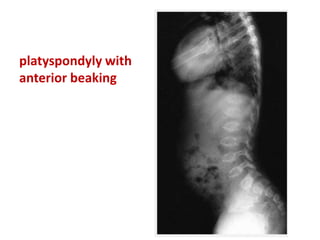
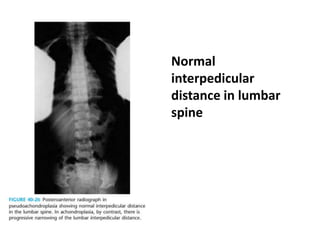
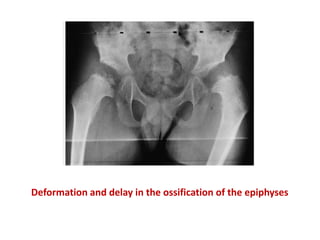
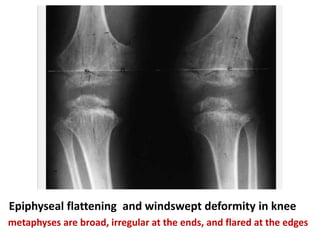
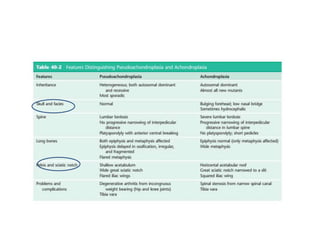
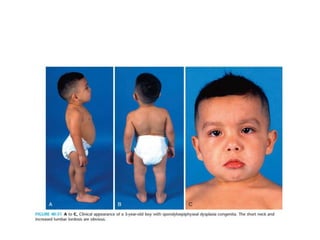
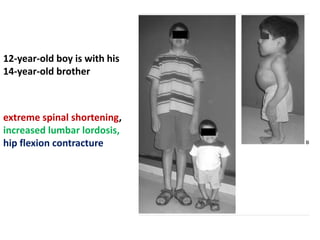
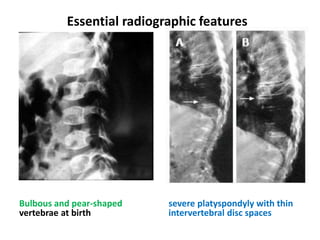
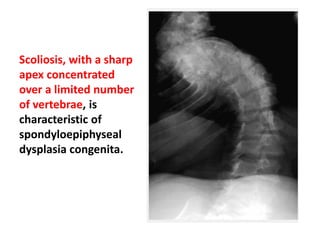
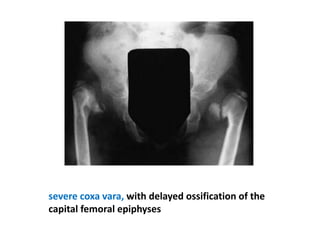
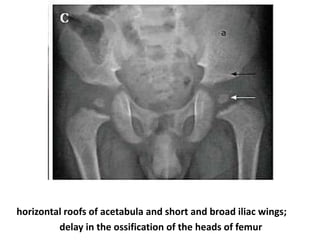
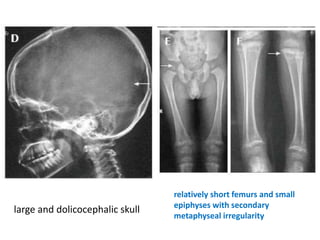
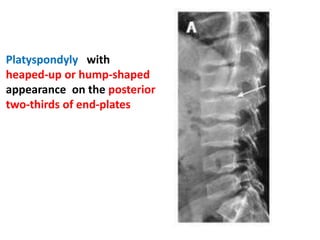
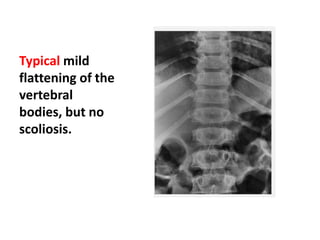
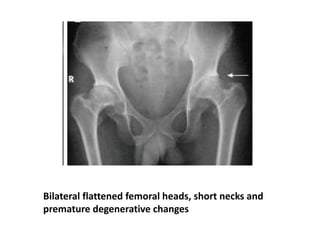
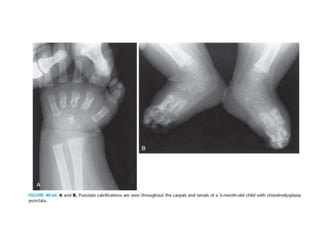
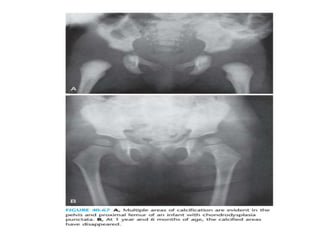
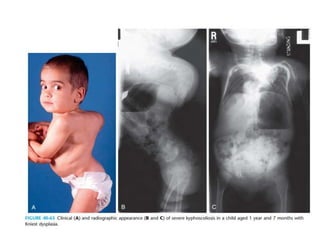
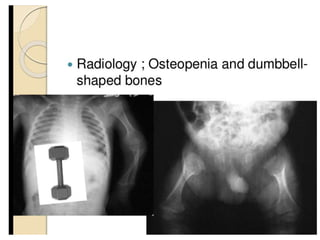
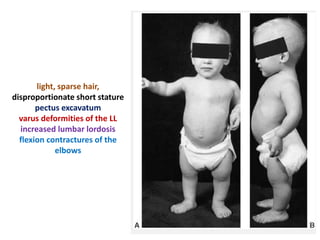
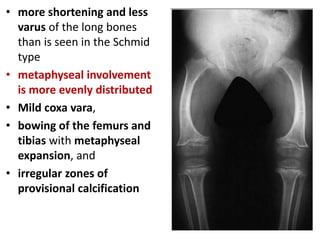
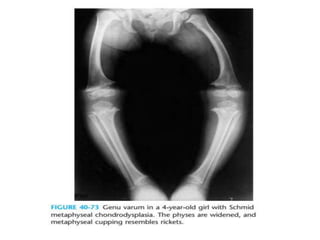
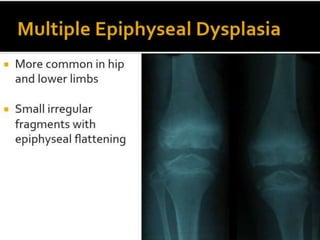
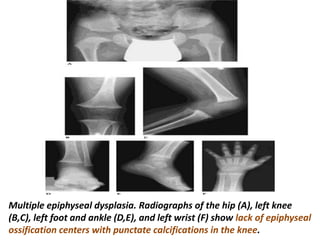
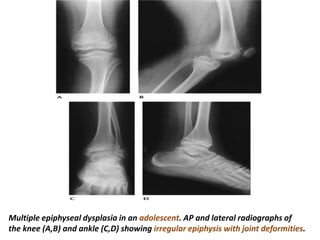
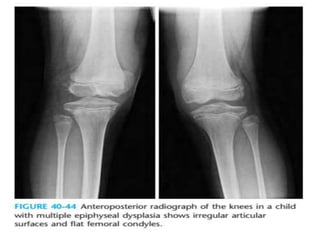
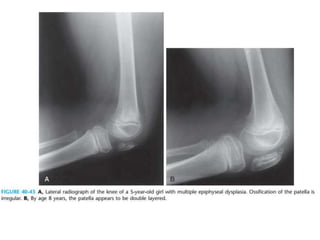
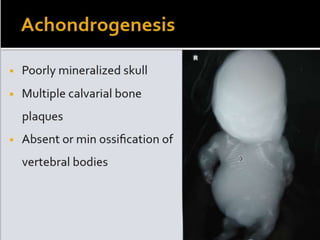
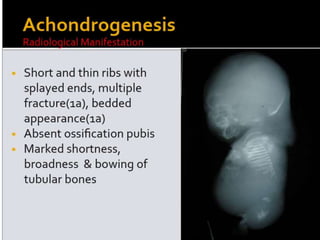
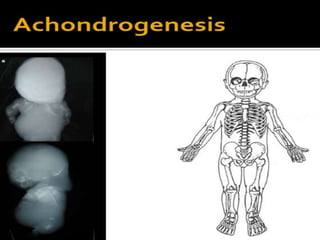
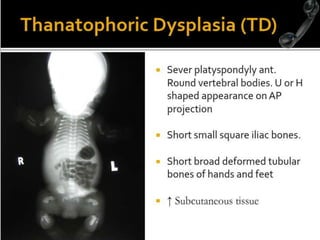
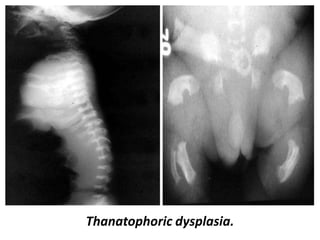
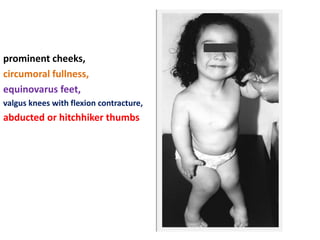
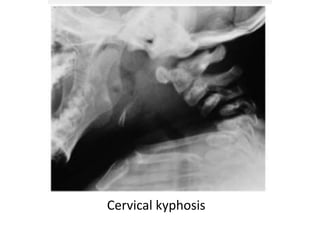
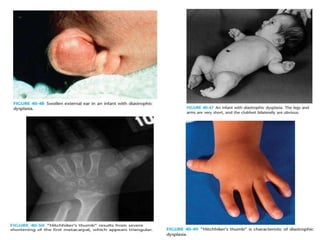
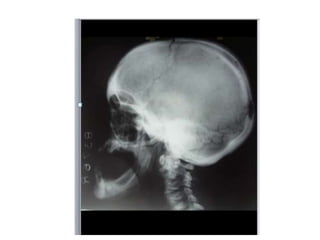
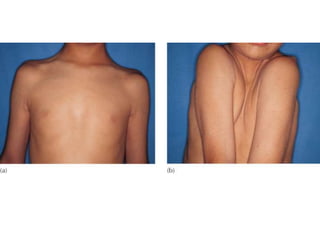
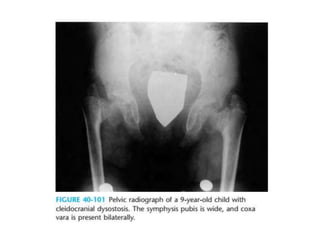
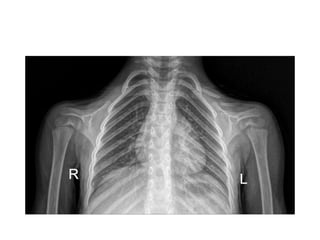
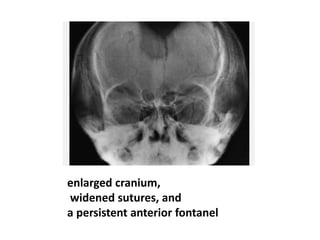
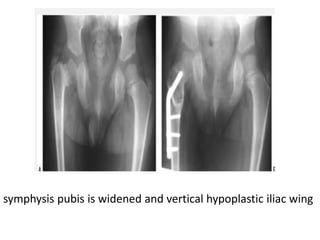
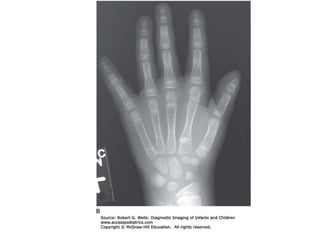
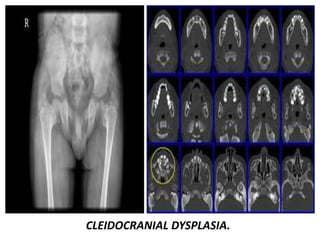
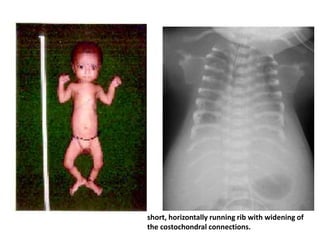
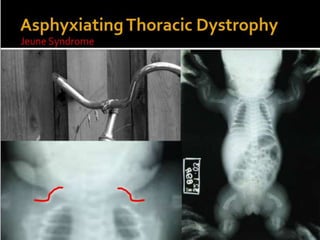
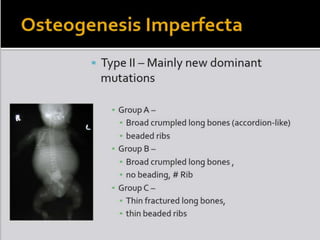
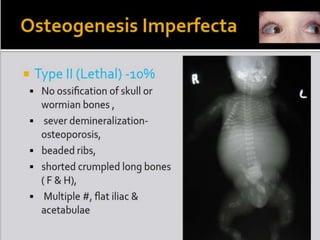
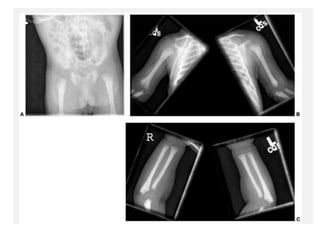
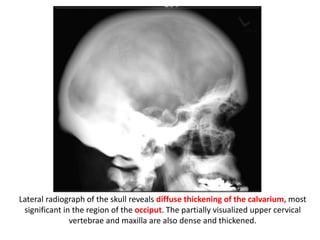
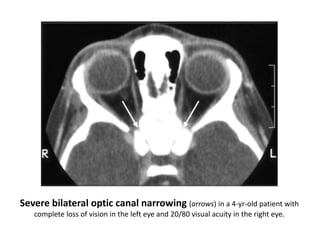
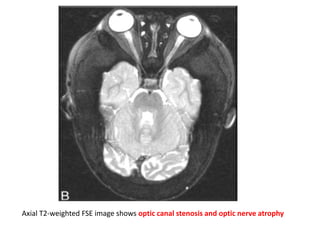
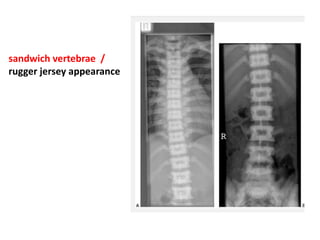
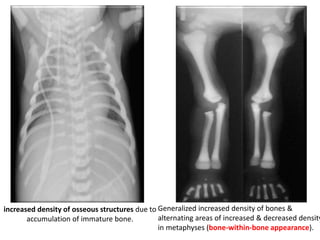
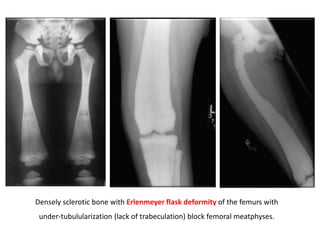
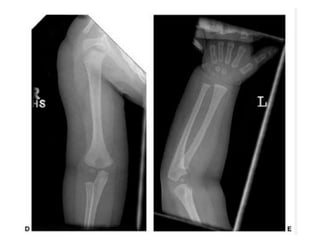
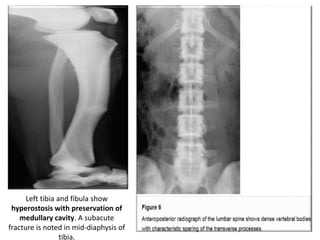
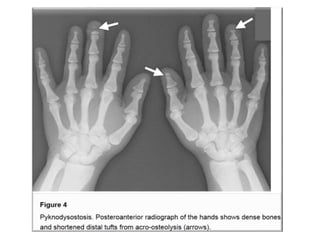
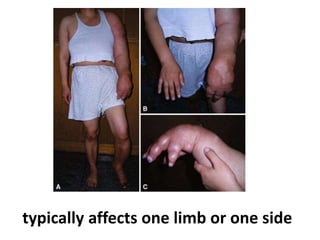
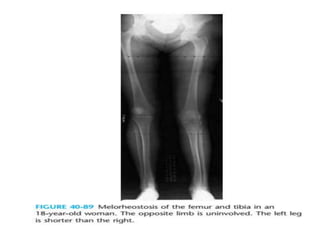
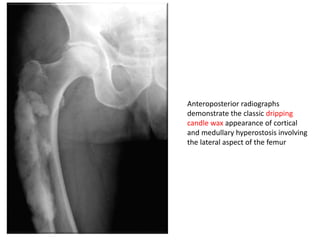
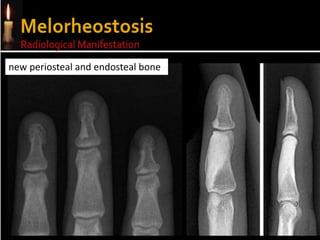
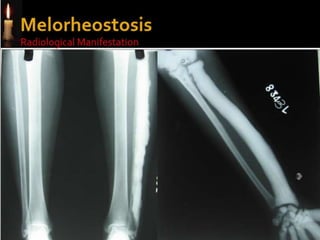
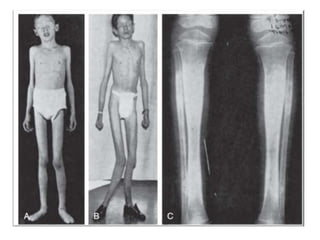
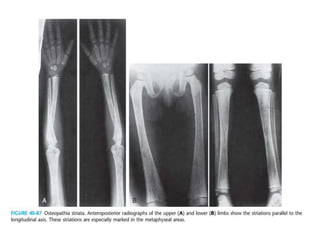
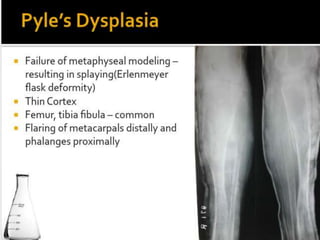
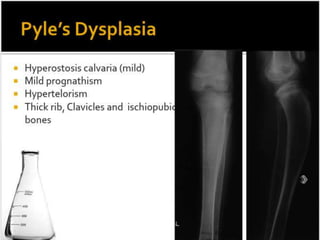
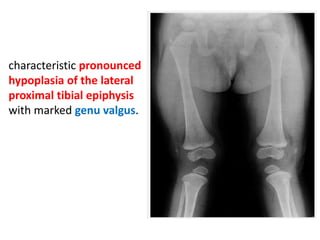
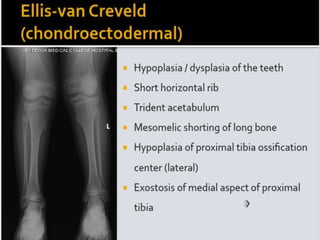
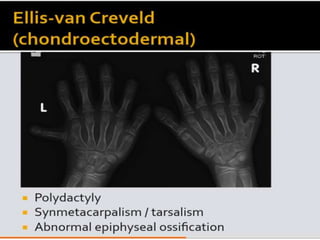
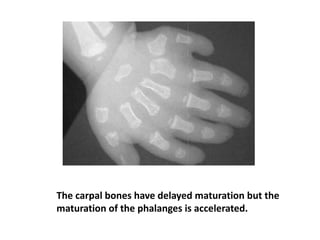
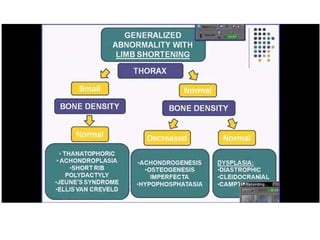
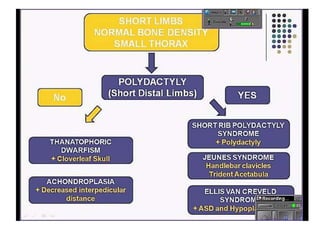
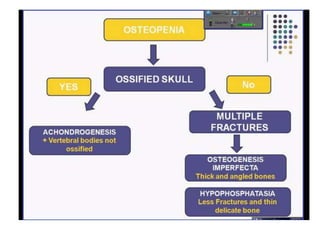
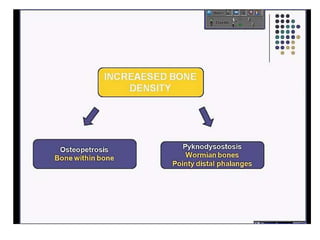
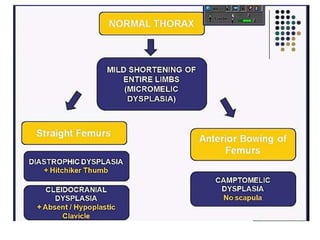
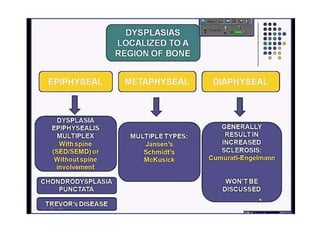
Skeletal dysplasias and dwarfism can be caused by over 200 genetic disorders that result in abnormal bone growth. There are two main types: dysplasias with generalized skeletal abnormalities and dysostoses with abnormalities of single or multiple bones. Achondroplasia, the most common type of dwarfism, is caused by a mutation that slows endochondral bone growth. It is characterized by frontal bossing, midface hypoplasia, rhizomelic shortening of the limbs, trident hands, genu varum, and normal intelligence. Assessment involves examining for disproportion, general examination including radiographs to evaluate for complications like foramen magnum stenosis. Treatment focuses on managing complications through surgery or other