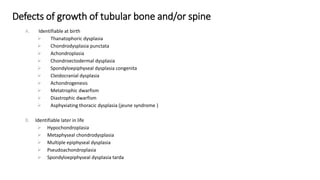
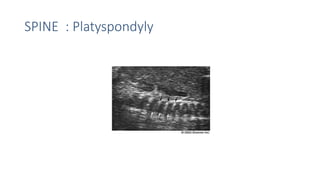
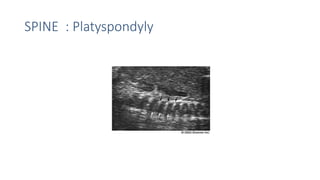
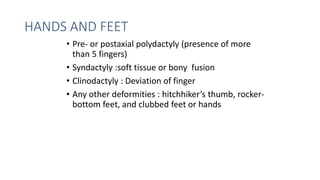
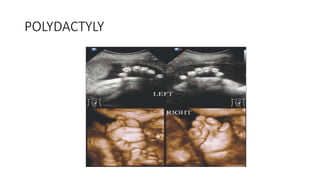
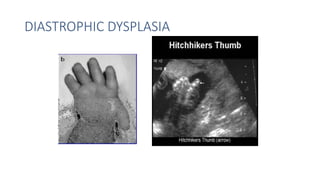
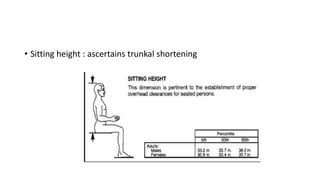
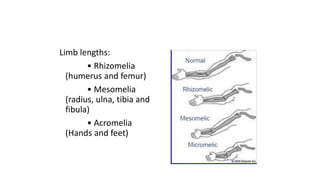
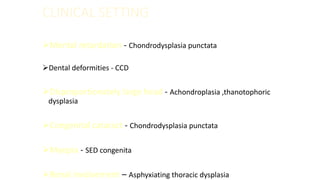
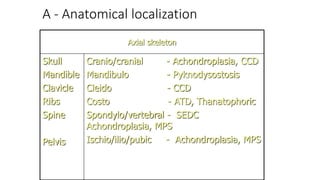
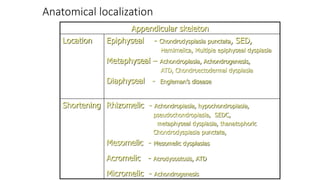
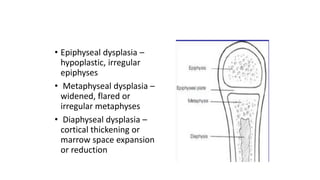
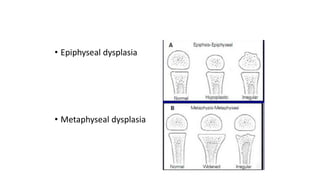
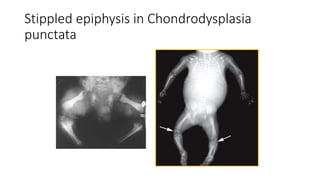
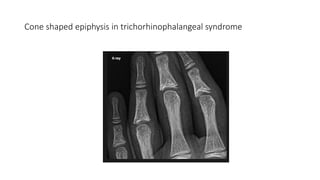
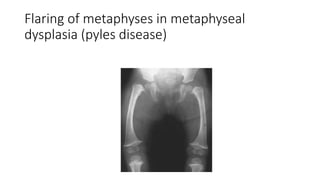
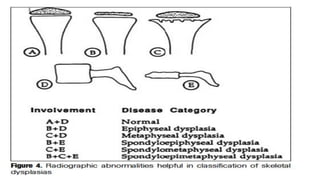
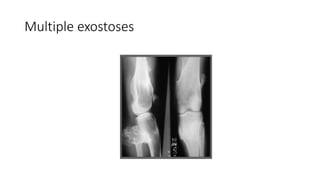
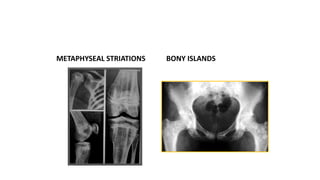
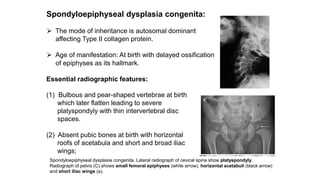
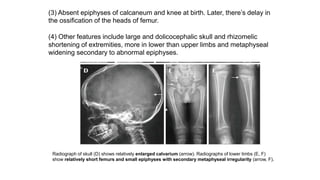
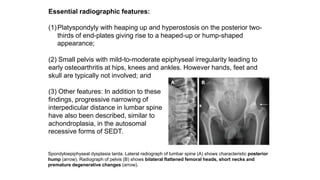
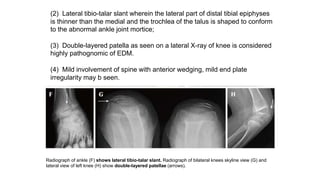
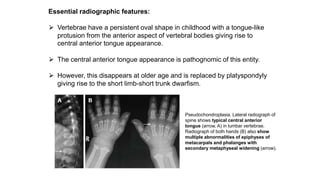
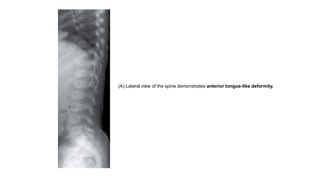
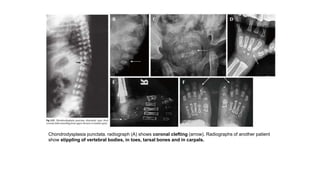
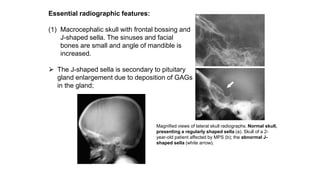
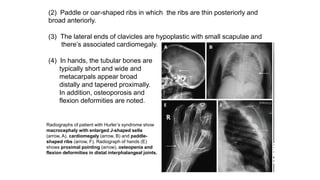
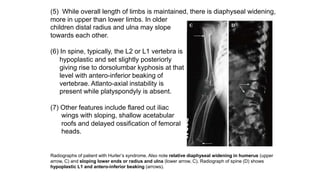
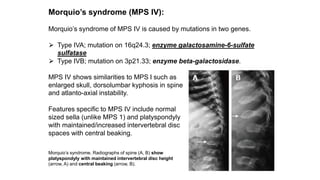
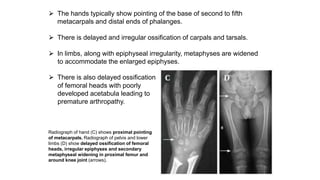
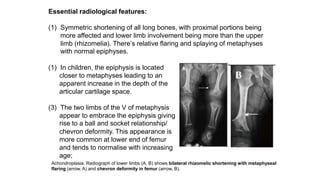
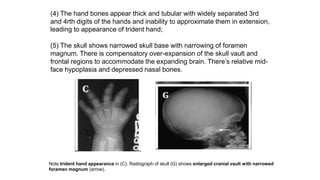
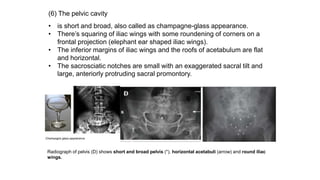
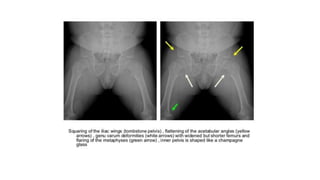
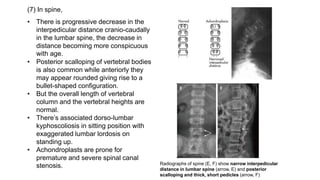
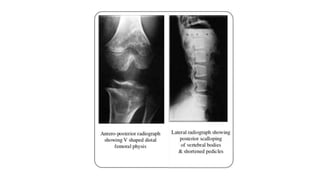
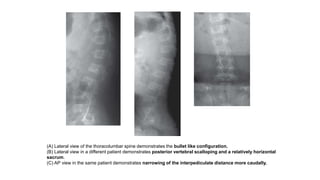
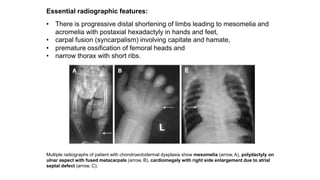
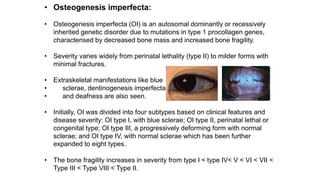
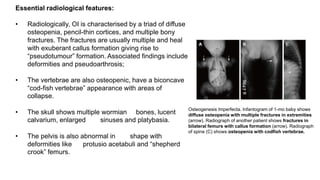
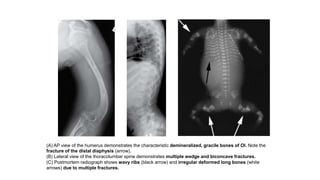
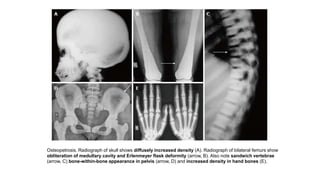
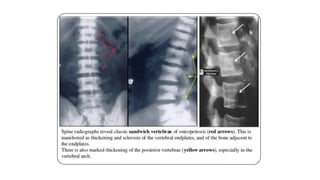
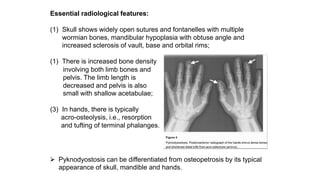
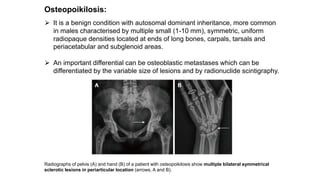
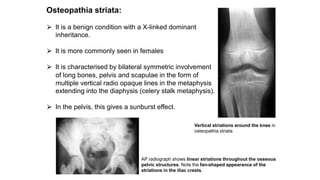
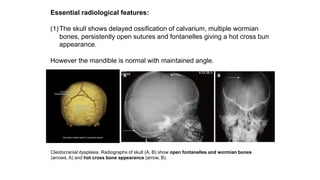
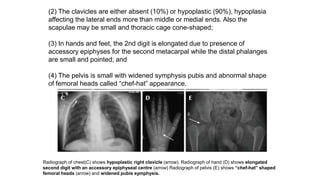
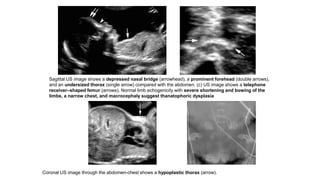
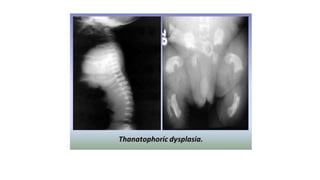
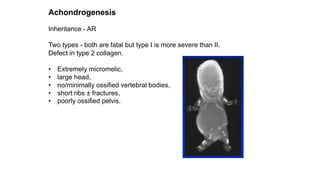
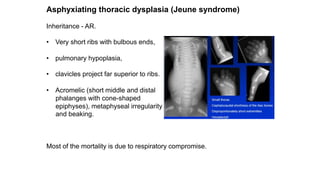
The document discusses skeletal dysplasias, a group of disorders affecting bone growth and development, characterized by abnormalities in shape, size, and density of bones. It classifies various conditions such as osteochondrodysplasias and deficiency of bone and cartilage growth, with a focus on prenatal and postnatal assessment methods including radiological evaluations. Additionally, it covers specific examples of skeletal dysplasias, their diagnosis, potential complications, and required interdisciplinary approaches for accurate diagnosis and management.