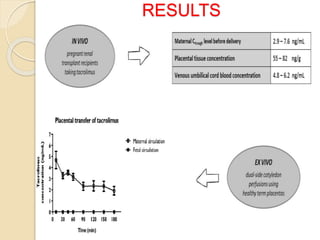
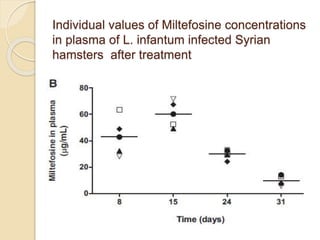
This document summarizes a seminar on computational methods for drug disposition. It discusses two approaches to modeling drug disposition: qualitative and quantitative. The quantitative approach uses pharmacophore modeling and docking to study drug interactions, while the qualitative approach uses QSAR and QSPR to correlate molecular descriptors with ADMET properties. The document also reviews the key processes of drug disposition: absorption, distribution, metabolism, and excretion. It provides examples of two research articles, one on the placental disposition of the immunosuppressant tacrolimus, and another on the pharmacokinetics of miltefosine in mice and hamsters infected with Leishmania.