

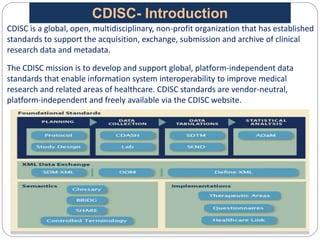

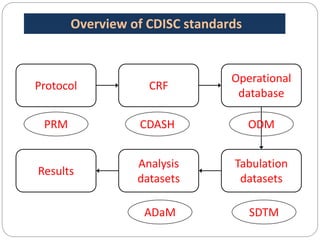










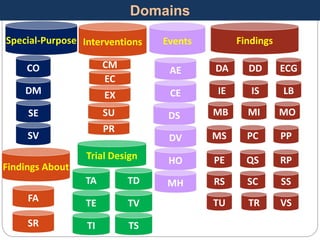



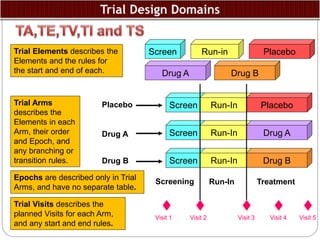

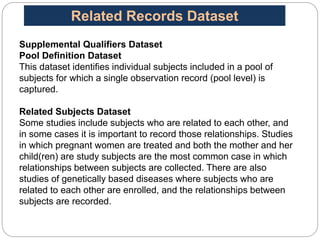






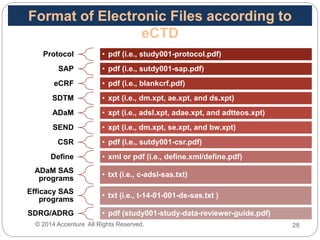






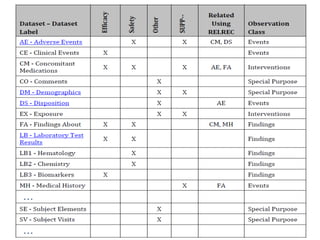
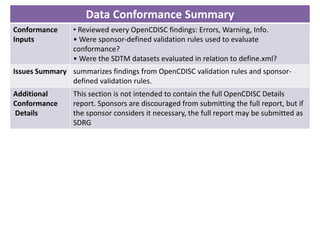



The Study Data Tabulation Model (SDTM), developed by CDISC, sets global standards for organizing clinical research data to facilitate regulatory submissions. It categorizes data into observations represented by domains based on events, findings, and interventions, with a focus on the structure and roles of variables collected during studies. Each dataset is supplemented by metadata that describes key attributes necessary for understanding the data in a regulatory context.





























































