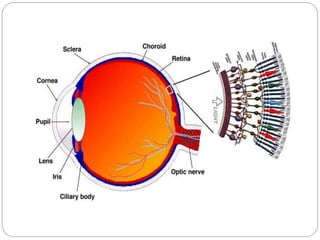
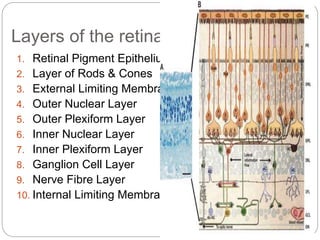
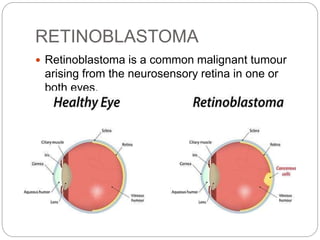
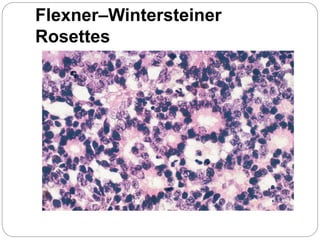
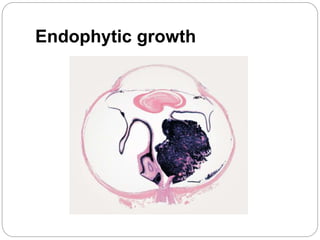
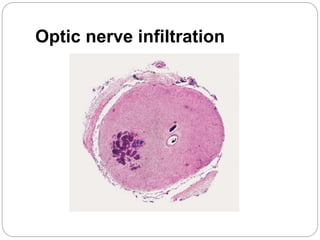
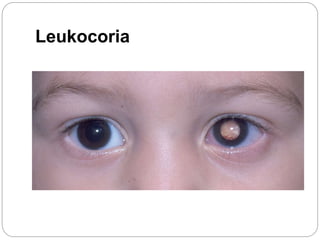
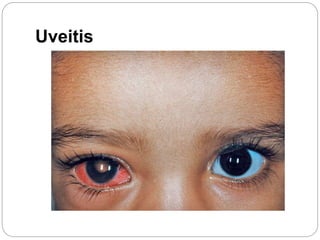
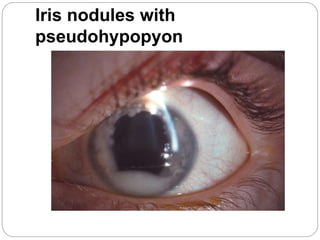
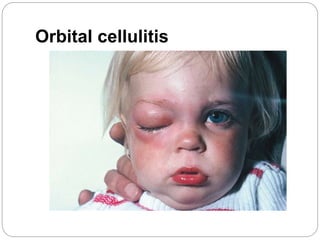
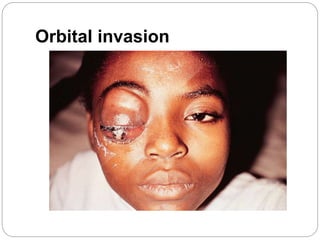
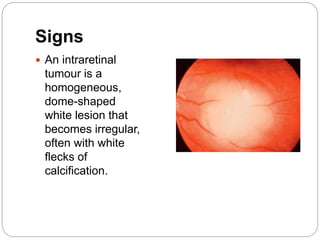
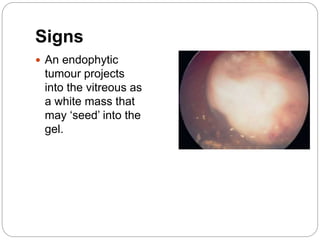
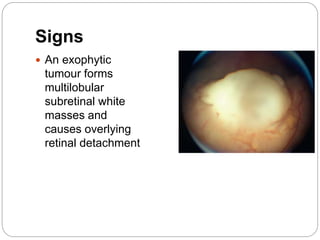
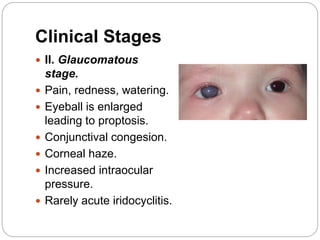
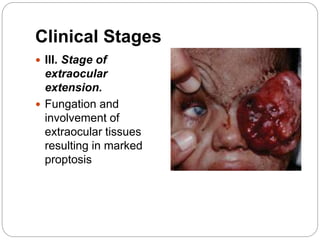
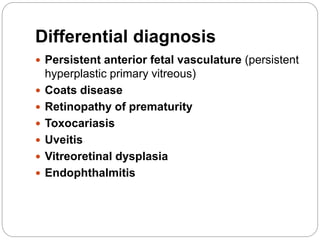
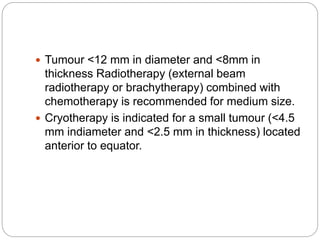
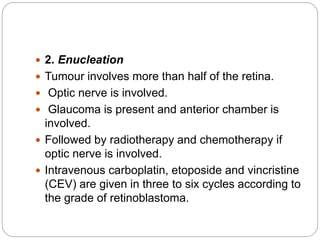
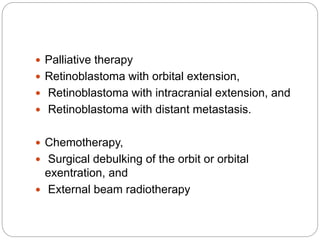
The retina is the innermost layer of the eye that contains photoreceptor cells. Retinoblastoma is a malignant tumor that arises from these photoreceptor cells in the retina, most commonly affecting young children under 5 years old. It can be hereditary if caused by a mutation in the RB1 gene, resulting in bilateral and multifocal tumors, or non-hereditary if caused by somatic mutations, usually presenting as a unilateral tumor. Treatment depends on tumor size and extent but may include chemotherapy, local therapies like cryotherapy or brachytherapy, and enucleation of the eye for advanced cases. Early diagnosis and treatment can help preserve vision and life.


















































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)












