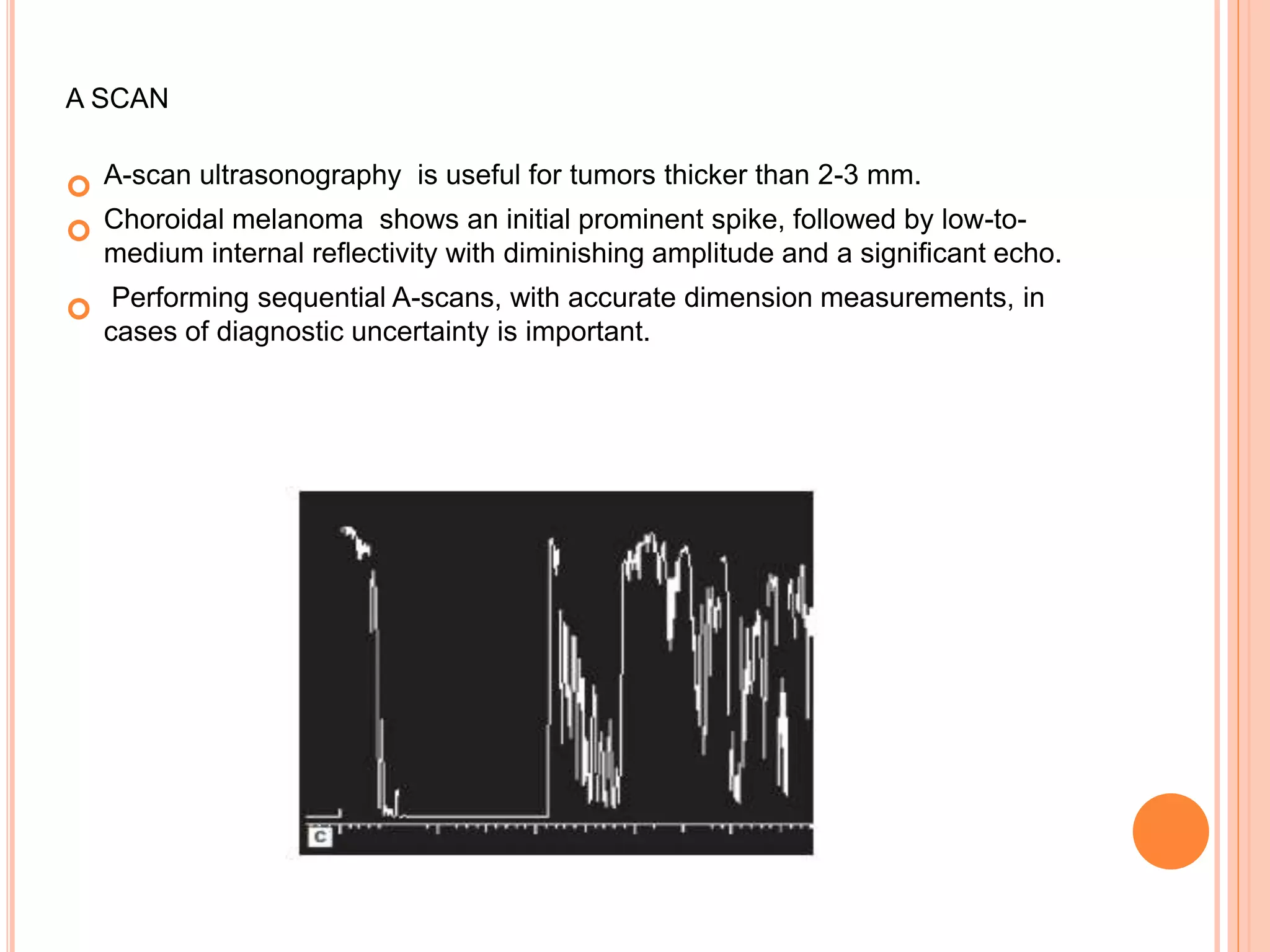
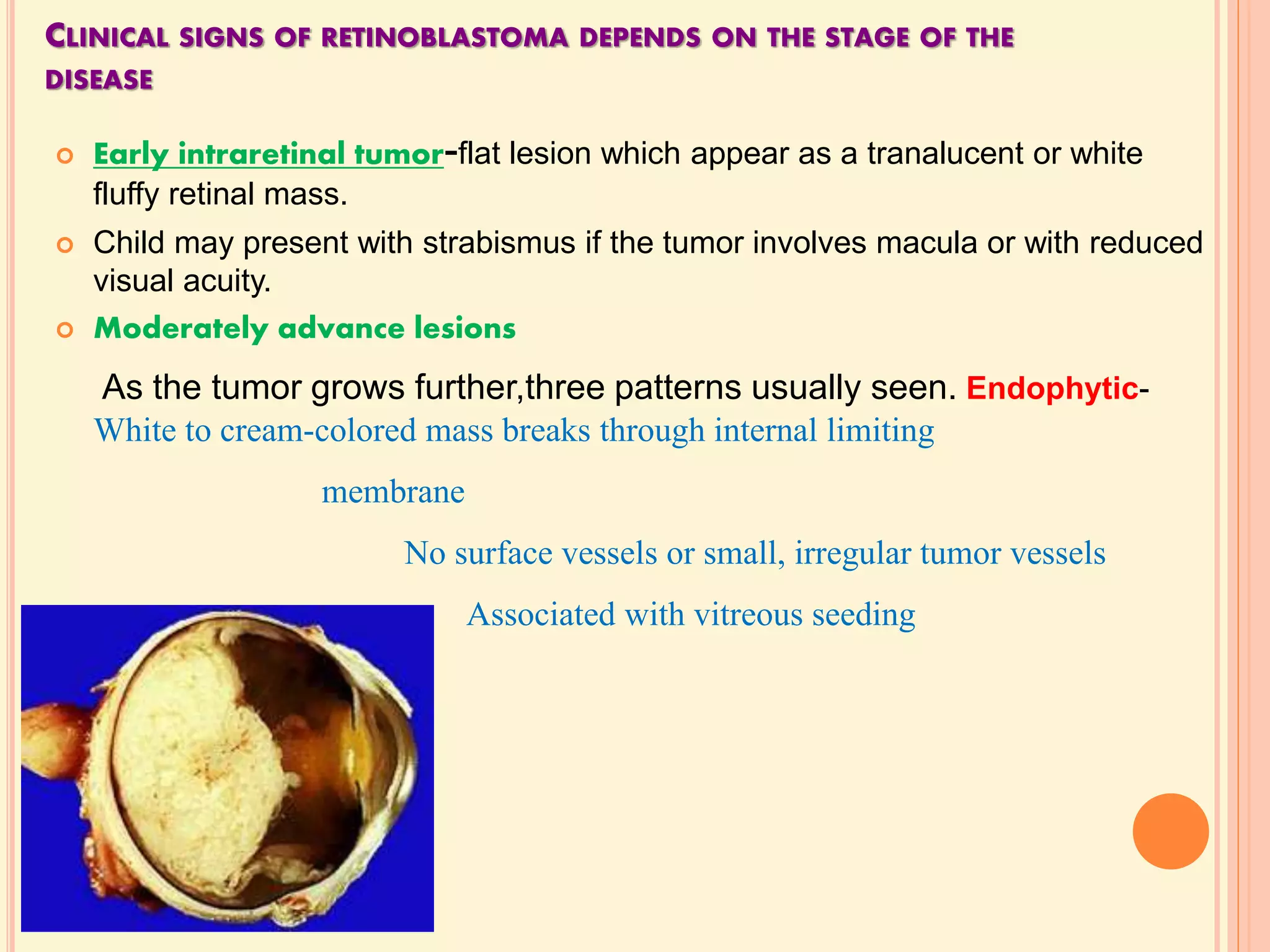
The document summarizes malignant intraocular tumors, focusing on retinoblastoma. It discusses retinoblastoma genetics, clinical features, diagnosis, staging, and treatment. Retinoblastoma is the most common intraocular malignancy in children, caused by deletion of the RB1 tumor suppressor gene. Clinical features include leukocoria. Diagnosis involves ophthalmic examination, imaging like CT/MRI, and histopathology showing Flexner-Wintersteiner rosettes. Staging systems include Reese-Ellsworth and the International Classification. Treatment depends on staging and includes focal therapies, chemotherapy, radiation, and enucleation. The goal is survival while retaining eyes and vision when possible
























![FOLLOW UP
1st 6 months: 4-6 week intervals
Upto 3 years: at 4-6 month intervals
Later, yearly
Family history positive: all family members [other children at
birth] should be examined yearly](https://image.slidesharecdn.com/intraoculartumors-191014062436/75/Intraocular-tumors-26-2048.jpg)