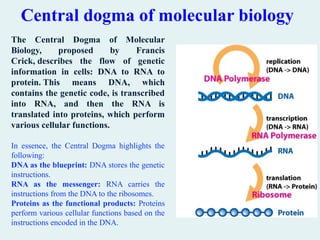
Central dogma ofmolecular biology
The Central Dogma of Molecular
Biology, proposed by Francis
Crick, describes the flow of genetic
information in cells: DNA to RNA to
protein. This means DNA, which
contains the genetic code, is transcribed
into RNA, and then the RNA is
translated into proteins, which perform
various cellular functions.
In essence, the Central Dogma highlights the
following:
DNA as the blueprint: DNA stores the genetic
instructions.
RNA as the messenger: RNA carries the
instructions from the DNA to the ribosomes.
Proteins as the functional products: Proteins
perform various cellular functions based on the
instructions encoded in the DNA.
3.
History of GeneticEngineering/
Recombinant DNA Technology
In conjunction with his studies of the tumor virus SV40, in 1972,
Paul Berg succeeded in inserting DNA from a bacterium into the
virus' DNA. He thereby created the first DNA molecule made
of parts from different organisms.
Paul Berg is the "father of genetic engineering/ rDNA Technology"
This type of molecule became known as
"hybrid DNA" or "recombinant DNA".
Among other things, Paul Berg's method
opened the way to creating bacteria that
produce substances used in medicines.
4.
History of Recombinant
DNATechnology
In 1973, Herbert Boyer, of the University of California at
San Francisco, and Stanley Cohen, at Stanford University,
reported the construction of functional organisms that
combined and replicated genetic information from different
species. Their experiments dramatically demonstrated the
potential impact of DNA recombinant engineering on medicine
and pharmacology, industry and agriculture.
Boyer and Cohen's achievement represented an advance upon
the ingenious techniques developed by Paul Berg, in 1972, for
inserting viral DNA into bacterial DNA. It was a creative
synthesis of earlier research that made use of:
Living organisms able to serve as carriers for genes from another organism.
Enzymes to cleave and rejoin DNA fragments that contain such genes.
DNA molecules from one organism precisely targeted and manipulated for
insertion into the DNA of another organism.
5.
Recombinant DNA and
GeneCloning
Recombinant DNA (rDNA) is a form of artificial
DNA that is created by combining two or more
sequences that would not normally occur together
through the process of gene splicing.
Recombinant DNA technology is a technology
which allows DNA to be produced via artificial
means. The procedure has been used to change
DNA in living organisms and may have even more
practical uses in the future.
6.
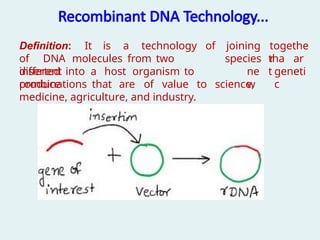
Recombinant DNA Technology...
togethe
r
Definition:It is a technology of
of DNA molecules from two
different
joining
species tha
t
ar
e
inserted into a host organism to
produce
ne
w
geneti
c
combinations that are of value to science,
medicine, agriculture, and industry.
What is RecombinantDNA Technology?
Recombinant DNA technology is a
technology which allows DNA to
be produced via artificial means.
The procedure has been used to change
DNA in living organisms and may have
even more practical uses in the future.
It is an area of medical science that is just
beginning to be researched in a
concerted effort.
9.
Recombinant DNAtechnology works by
taking DNA from two different sources and
combining that DNA into a single molecule.
That alone, however, will not do much.
Recombinant DNA technology only
becomes useful when that artificially-
created DNA is reproduced. This is known
as DNA cloning.
Recombinant DNA Technology
1.The basic concepts for recombinant
DNA technology
2. The basic procedures of recombinant
DNA technology
3. Application of recombinant DNA
technology
In theearly 1970s, technologies for the
laboratory manipulation of nucleic acids
emerged. In turn, these technologies led
to the construction of DNA molecules
composed of nucleotide sequences taken
from different sources. The products of
these innovations, recombinant DNA
molecules, opened exciting new avenues
of investigation in molecular biology and
genetics, and a new field was born—
recombinant DNA technology.
14.
Concept of RecombinantDNA
Recombinant DNA is a molecule that combines
DNA from two sources . Also known as gene
cloning.
Creates a new combination of genetic material
– Human gene for insulin was placed in bacteria
– The bacteria are recombinant organisms and
produce insulin in large quantities for diabetics
– Genetically engineered drug in 1986
Genetically modified organisms are possible
because of the universal nature of the genetic
code!
15.
Genetic engineeringis the application
of this technology to the
manipulation of genes. These
advances were made possible by
methods for amplification of any
particular DNA segment( how? ),
regardless of source, within bacterial
host cells. Or, in the language of
recombinant DNA technology, the
cloning of virtually any DNA sequence
became feasible.
16.
Recombinant technologybegins with the
isolation of a gene of interest (target gene).
The target gene is then inserted into the
plasmid or phage (vector) to form replicon.
The replicon is then introduced into host cells
to cloned and either express the protein or not.
The cloned replicon is referred to as
recombinant DNA. The procedure is called
recombinant DNA technology. Cloning is
necessary to produce numerous copies of the
DNA since the initial supply is inadequate to
insert into host cells.
17.
Some otherterms are also in common use to
describe genetic engineering.
Gene manipulation
Recombinant DNA technology
Gene cloning (Molecular cloning)
Genetic modification
18.
Cloning——In classicalbiology, a clone is a
population of identical organisms derived
from a single parental organism.
For example, the members of a colony of
bacterial cells that arise from a single cell on a
petri plate are clones. Molecular biology
has borrowed the term to mean a collection
of molecules or cells all identical to an
original molecule or cell.
19.
How recombinant technologyworks
These steps include isolating of the target
gene and the vector, specific cutting of
DNA at defined sites, joining or splicing of
DNA fragments, transforming of
replicon to host cell, cloning, selecting of
the positive cells containing
recombinant DNA, and either express or
not in the end.
DNA moleculesthat are constructed with DNA
from different sources are called recombinant
DNA molecules.
Recombinant DNA molecules are created
in nature more often than in the laboratory;
– for example, every time a bacteria phage or
eukaryotic virus infects its host cell and
integrates its DNA into the host genome, a
recombinant is created.
– Occasionally, these viruses pick up a fragment
of host DNA when they excise from their
host’s genome; these naturally occurring
recombinant DNA molecules have been
used to study some genes.
24.
Six basic stepsare common to most
recombinant DNA experiments
1. Isolation and purification of DNA.
Both vector and target DNA
molecules can be prepared by a
variety of routine methods, which are
not discussed here. In some cases, the
target DNA is synthesized in vitro.
25.
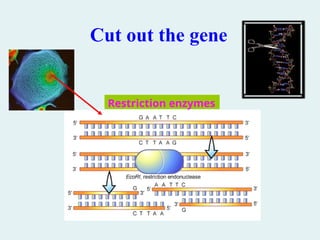
2. Cleavage ofDNA at particular sequences. As
we will see, cleaving DNA to generate
fragments of defined length, or with specific
endpoints, is crucial to recombinant DNA
technology. The DNA fragment of interest is
called insert DNA. In the laboratory, DNA is
usually cleaved by treating it with
commercially produced nucleases and
restriction endonucleases.
26.
3. Ligation ofDNA fragments.
A recombinant DNA molecule is usually
formed by cleaving the DNA of interest to
yield insert DNA and then ligating the
insert DNA to vector DNA (recombinant
DNA or chimeric DNA). DNA fragments are
typically joined using DNA ligase (also
commercially produced).
– T4 DNA Ligase
27.
4. Introduction ofrecombinant DNA into
compatible host cells. In order to be
propagated, the recombinant DNA
molecule (insert DNA joined to vector
DNA) must be introduced into a
compatible host cell where it can replicate.
The direct uptake of foreign DNA by a host
cell is called genetic transformation (or
transformation). Recombinant DNA can
also be packaged into virus particles and
transferred to host cells by transfection.
28.
5. Replication andexpression of
recombinant DNA in host cells.
Cloning vectors allow insert DNA to be
replicated and, in some cases, expressed
in a host cell. The ability to clone and
express DNA efficiently depends on the
choice of appropriate vectors and hosts.
29.
6. Identification ofhost cells that contain
recombinant DNA of interest. Vectors
usually contain easily scored genetic
markers, or genes, that allow the
selection of host cells that have taken up
foreign DNA. The identification of a
particular DNA fragment usually
involves an additional step—screening a
large number of recombinant DNA
clones. This is almost always the most
difficult step.
30.
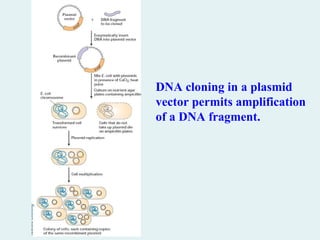
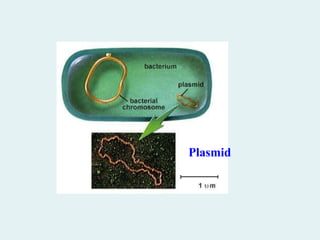
DNA cloning ina plasmid
vector permits amplification
of a DNA fragment.
How to geta target genes?
1. Genomic DNA
2. Artificial synthesis
3. PCR amplification
4. RT-PCR
33.
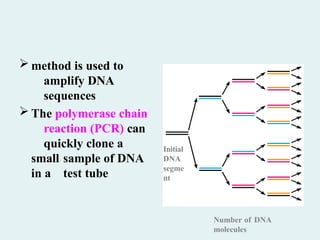
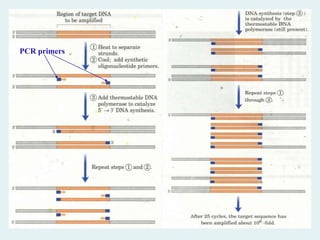
Polymerase chain reaction(PCR)
A technique called the polymerase chain
reaction (PCR) has revolutionized
recombinant DNA technology. It can
amplify DNA from as little material as
a single cell and from very old tissue
such as that isolated from Egyptian
mummies, a frozen mammoth, and
insects trapped in ancient amber.
34.
method isused to
amplify DNA
sequences
The polymerase chain
reaction (PCR) can
quickly clone a
small sample of DNA
in a test tube
Number of DNA
molecules
Initial
DNA
segme
nt
RT-PCR
Reverse transcriptionpolymerase chain reaction
(RT-PCR) is a variant of polymerase chain
reaction (PCR.
In RT-PCR, however, an RNA strand is first
reverse transcribed into its DNA complement
(complementary DNA, or cDNA) using the enzyme
reverse transcriptase, and the resulting cDNA is
amplified using traditional.
– Template:RNA
– Products: cDNA
37.
Vectors- Cloning Vehicles
Cloningvectors can be plasmids,
bacteriophage, viruses, or even small
artificial chromosomes. Most vectors
contain sequences that allow them to be
replicated autonomously within a
compatible host cell, whereas a minority
carry sequences that facilitate integration
into the host genome.
38.
All cloningvectors have in common at least
one unique cloning site, a sequence that
can be cut by a restriction endonuclease to
allow site-specific insertion of foreign
DNA. The most useful vectors have
several restriction sites grouped together
in a multiple cloning site (MCS) called a
polylinker.
39.
Types of vector
1.Plasmid Vectors
2. Bacteriophage Vectors
3. Virus vectors
4. Shuttle Vectors--can replicate in either
prokaryotic or eukaryotic cells.
5. Yeast Artificial Chromosomes as
Vectors
40.
Plasmid Vectors
Plasmidsare circular, double-stranded
DNA (dsDNA) molecules that are separate
from a cell’s chromosomal DNA.
These extra chromosomal DNAs, which
occur naturally in bacteria and in lower
eukaryotic cells (e.g., yeast), exist in a
parasitic or symbiotic relationship with
their host cell.
Plasmids canreplicate autonomously within
a host, and they frequently carry genes
conferring resistance to antibiotics such as
tetracycline, ampicillin, or kanamycin.
The expression of these marker genes can
be used to distinguish between host cells
that carry the vectors and those that do not
43.
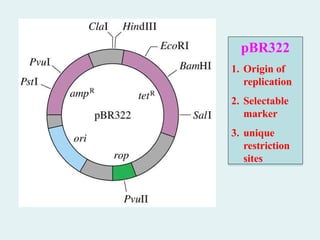
pBR322
pBR322 wasone of the first versatile plasmid
vectors developed; it is the ancestor of many of the
common plasmid vectors used in biochemistry
laboratories.
pBR322 contains an origin of replication (ori) and
a gene (rop) that helps regulate the number of
copies of plasmid DNA in the cell. There are two
marker genes: confers resistance to ampicillin,
and confers resistance to tetracycline. pBR322
contains a number of unique restriction sites that
are useful for constructing recombinant DNA.
Enzymes
Restriction Enzymes andDNA Ligases Allow
Insertion of DNA Fragments into Cloning Vectors
1. Restriction endonuclease, RE
2. DNA ligase
3. Reverse transcriptase
4. DNA polymerase, DNA pol
5. Nuclease
6. Terminal transferase
46.
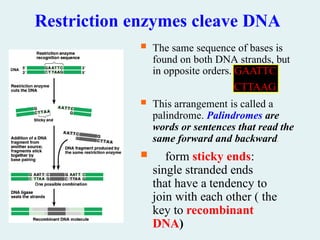
Restriction enzymes cleaveDNA
The same sequence of bases is
found on both DNA strands, but
in opposite orders. GAATTC
CTTAAG
This arrangement is called a
palindrome. Palindromes are
words or sentences that read the
same forward and backward.
form sticky ends:
single stranded ends
that have a tendency to
join with each other ( the
key to recombinant
DNA)
47.
Restriction Enzymes CutDNA Chains at
Specific Locations
Restriction enzymes are endonucleases
produced by bacteria that typically
recognize specific 4 to 8bp sequences,
called restriction sites, and then cleave both
DNA strands at this site.
Restriction sites commonly are short
palindromic sequences; that is, the
restriction-site sequence is the same on
each DNA strand when read in the 5′ → 3′
direction.
Restriction enzymes
Restrictionenzymes are named after the
bacterium from which they are isolated
– For example, Eco RI is from Escherichia
coli, and Bam HI is from Bacillus
amyloliquefaciens . The first three letters in the
restriction enzyme name consist of the first
letter of the genus (E) and the first two letters of
the species (co). These may be followed by a
strain designation (R) and a roman numeral (I)
to indicate the order of discovery (eg, EcoRI,
EcoRII).
50.
Blunt ends orsticky ends
Each enzyme recognizes and cleaves a
specific double-stranded DNA sequence that
is 4–7 bp long. These DNA cuts result in
blunt ends (eg, Hpa I) or overlapping
(sticky) ends (eg, BamH I) , depending on the
mechanism used by the enzyme.
Sticky ends are particularly useful in
constructing hybrid or chimeric
DNA molecules .
51.
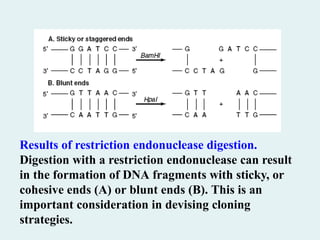
Results of restrictionendonuclease digestion.
Digestion with a restriction endonuclease can result
in the formation of DNA fragments with sticky, or
cohesive ends (A) or blunt ends (B). This is an
important consideration in devising cloning
strategies.
53.
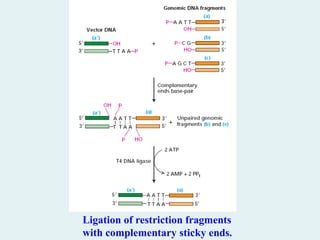
Inserting DNA Fragmentsinto Vectors
DNA fragments with either sticky ends or blunt
ends can be inserted into vector DNA with the
aid of DNA ligases.
For purposes of DNA cloning, purified DNA
ligase is used to covalently join the ends of a
restriction fragment and vector DNA that have
complementary ends . The vector DNA and
restriction fragment are covalently ligated
together through the standard 3 → 5
phosphodiester bonds of DNA.
DNA ligase “pastes” the DNA fragments
together
Identification of HostCells
Containing Recombinant DNA
Once a cloning vector and insert DNA have
been joined in vitro, the recombinant DNA
molecule can be introduced into a host cell,
most often a bacterial cell such as E. coli.
In general, transformation is not a very
efficient way of getting DNA into a cell
because only a very small percentage of cells
take up recombinant DNA. Consequently,
those cells that have been successfully
transformed must be distinguished from the
vast majority of untransformed cells.
56.
Identification ofhost cells containing
recombinant DNA requires genetic selection or
screening or both.
In a selection, cells are grown under conditions in
which only transformed cells can survive; all the
other cells die.
In contrast, in a screen, transformed cells have to
be individually tested for the presence of the
desired recombinant DNA.
Normally, a number of colonies of cells are
first selected and then screened for colonies
carrying the desired insert.
57.
Selection Strategies UseMarker Genes
(Primary screening)
Many selection strategies involve selectable
marker genes— genes whose presence
can easily be detected or demonstrated.
ampR
Selection or screening can also be achieved
using insertional inactivation.
58.
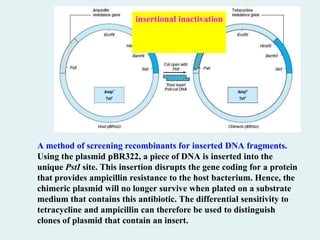
A method ofscreening recombinants for inserted DNA fragments.
Using the plasmid pBR322, a piece of DNA is inserted into the
unique PstI site. This insertion disrupts the gene coding for a protein
that provides ampicillin resistance to the host bacterium. Hence, the
chimeric plasmid will no longer survive when plated on a substrate
medium that contains this antibiotic. The differential sensitivity to
tetracycline and ampicillin can therefore be used to distinguish
clones of plasmid that contain an insert.
insertional inactivation
59.
Screening (Strategies)
1. GelElectrophoresis Allows Separation of
Vector DNA from Cloned Fragments
2. Cloned DNA Molecules Are Sequenced
Rapidly by the Dideoxy Chain-Termination
Method
3. The Polymerase Chain Reaction Amplifies a
Specific DNA Sequence from a Complex
Mixture
4. Blotting Techniques Permit Detection of
Specific DNA Fragments and mRNAs with
DNA Probes
60.
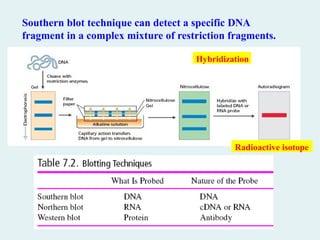
Southern blot techniquecan detect a specific DNA
fragment in a complex mixture of restriction fragments.
Radioactive isotope
Hybridization
61.
Types of blottingtechniques
Southern blotting
Southern blotting techniques is the first nucleic acid
blotting procedure developed in 1975 by
Southern.
Southern blotting is the techniques for the specific
identification of DNA molecules.
Northern blotting
Northern blotting is the techniques for the specific
identification of RNA molecules.
Western blotting
Western blotting involves the identification of
proteins.
Antigen + antibody
62.
Expression of ProteinsUsing
Recombinant DNA Technology
Cloned or amplified DNA can be purified and
sequenced, used to produce RNA and protein, or
introduced into organisms with the goal of
changing their phenotype.
One of the reasons recombinant DNA technology
has had such a large impact on biochemistry is
that it has overcome many of the difficulties
inherent in purifying low-abundance proteins and
determining their amino acid sequences.
63.
Recombinant DNAtechnology allows the
protein to be purified without further
characterization. Purification begins
with overproduction of the protein in a
cell containing an expression vector.
– Prokaryotic Expression Vectors
– Eukaryotic Expression Vectors
64.
Prokaryotic Expression Vectors
Expression vectors for bacterial hosts are
generally plasmids that have been
engineered to contain appropriate
regulatory sequences for transcription and
translation such as strong promoters,
ribosome-binding sites, and transcription
terminators.
65.
Eukaryotic proteinscan be made in bacteria by
inserting a cDNA fragment into an expression
vector . Large amounts of a desired protein can be
purified from the transformed cells.
In some cases, the proteins can be used to treat
patients with genetic disorders.
For example, human growth hormone, insulin, and
several blood coagulation factors have been
produced using recombinant DNA technology
and expression vectors.
66.
Expression of Proteinsin Eukaryotes
Prokaryotic cells may be unable to produce
functional proteins from eukaryotic
genes even when all the signals
necessary for gene expression are present
because many eukaryotic proteins must
be post- translationally modified.
67.
Several expressionvectors that function in
eukaryotes have been developed.
These vectors contain eukaryotic origins of
replication, marker genes for selection in
eukaryotes, transcription and translation
control regions, and additional features
required for efficient translation of
eukaryotic mRNA, such as polyadenylation
signals and capping sites.