














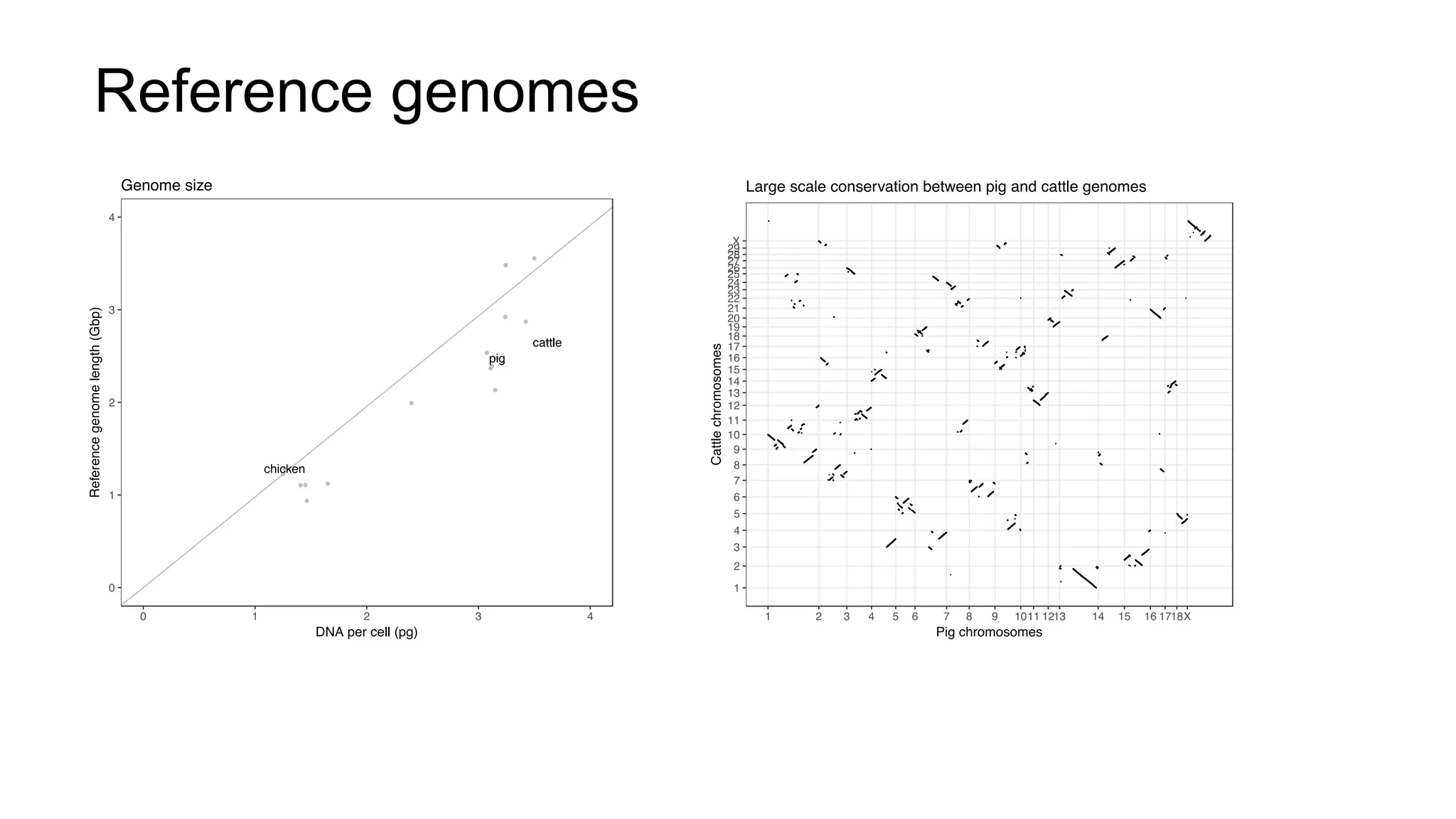


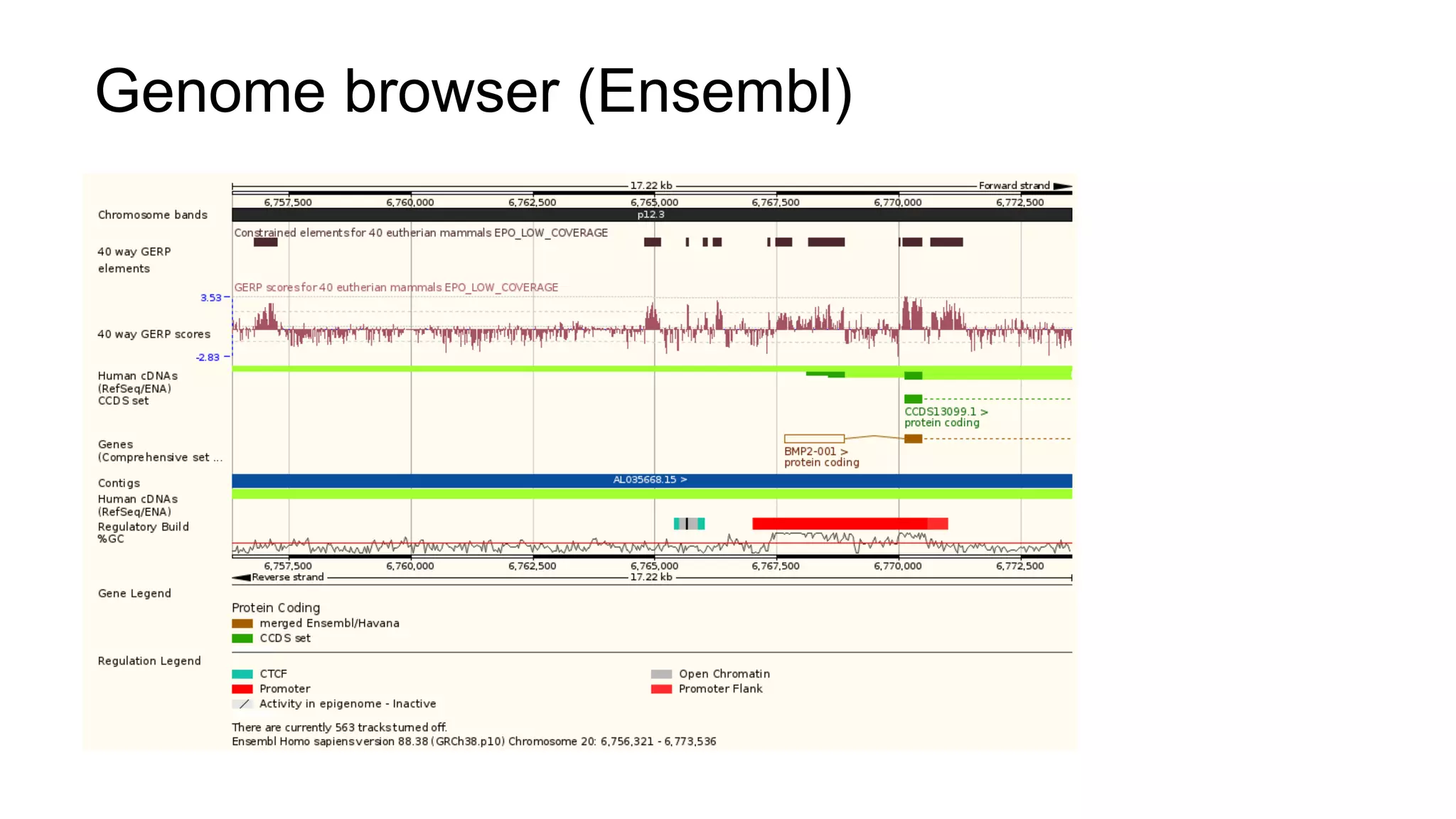











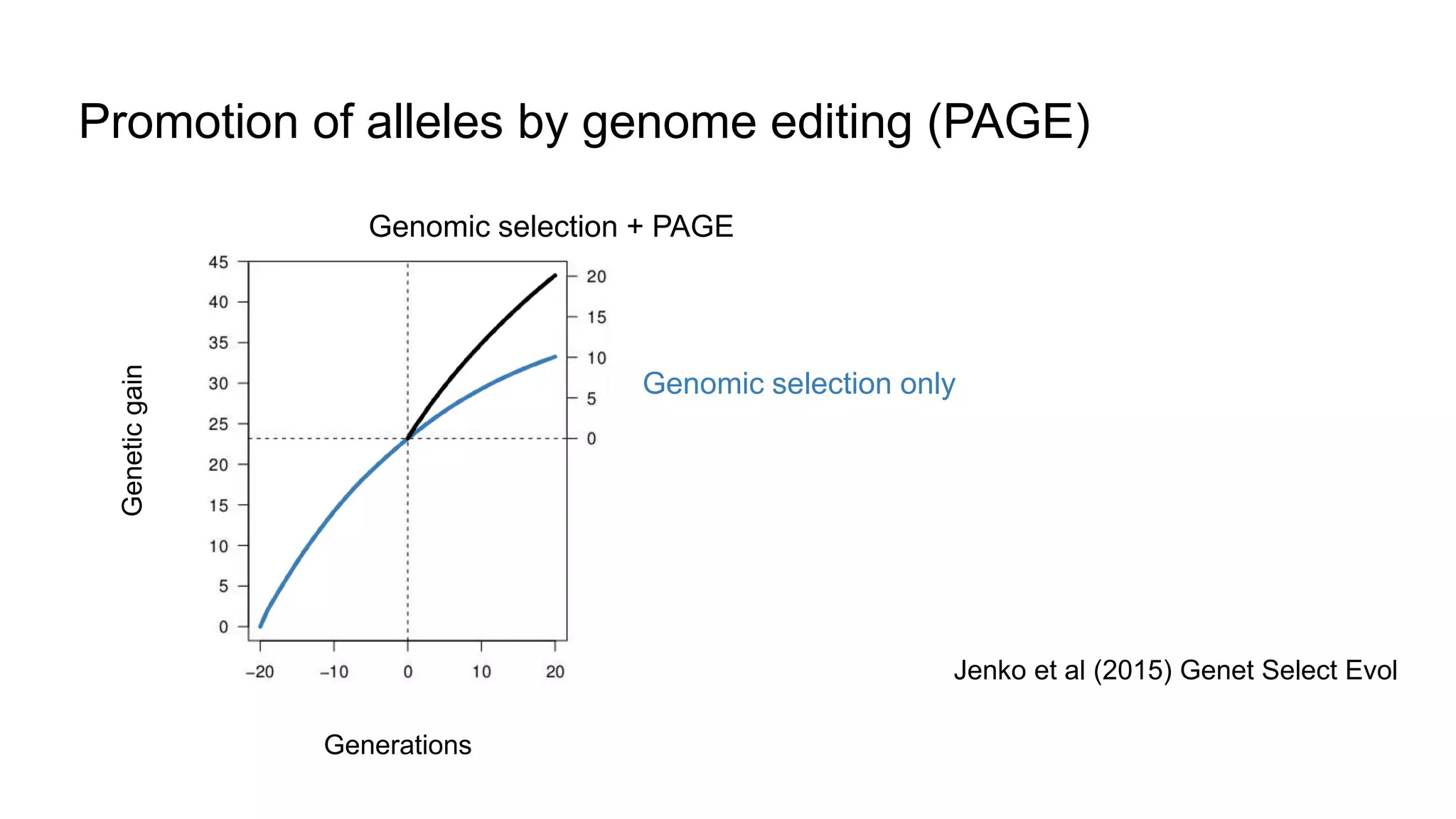
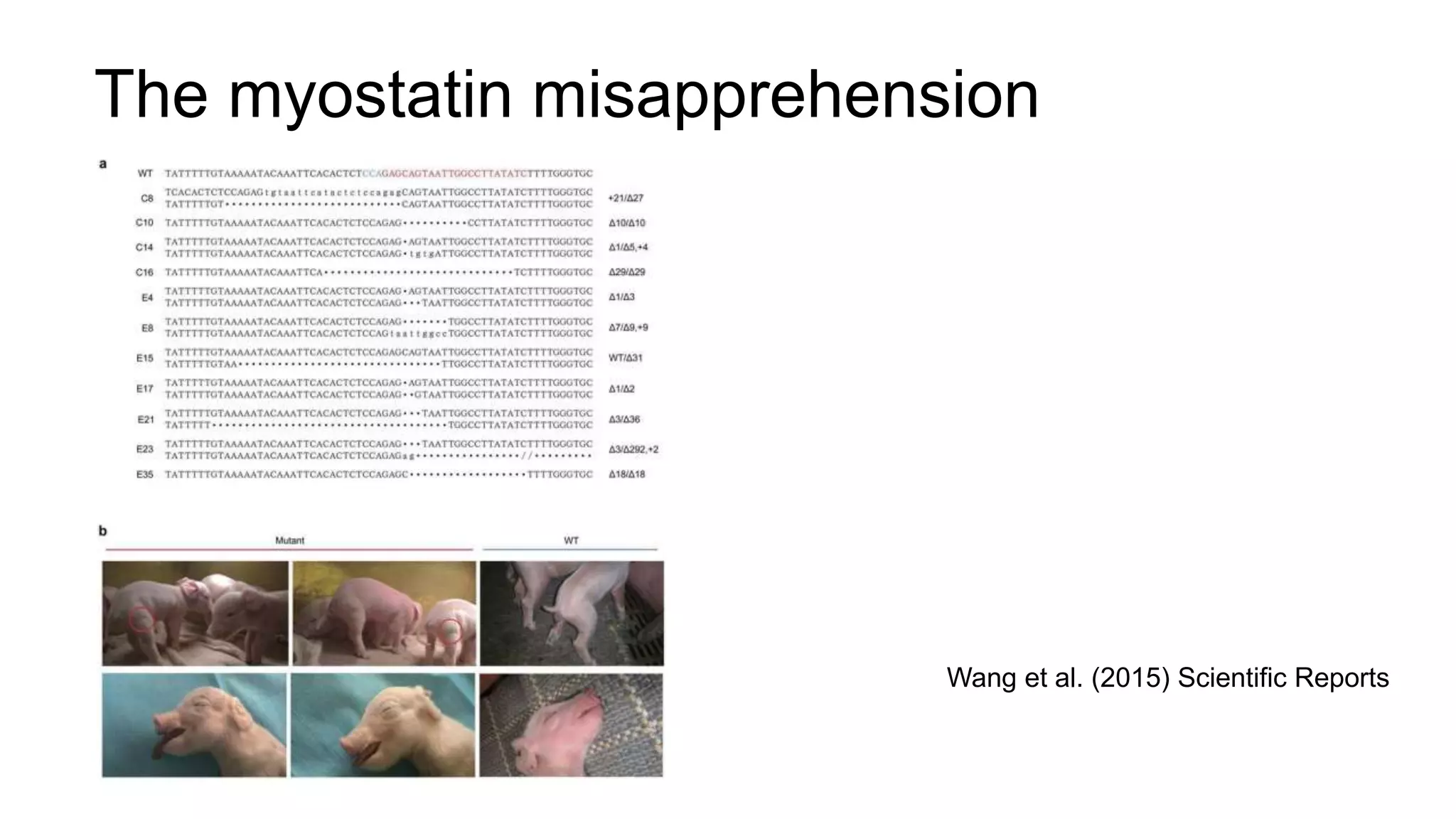






The document discusses the integration of molecular genetics with traditional agricultural methods to enhance the economic value of domesticated species. It highlights advancements in genomic selection and the development of genetic tools that improve the accuracy and efficiency of breeding programs. The future of animal breeding is expected to see significant changes with advancements in gene function discovery and the incorporation of DNA-based tests into breeding practices.



































































