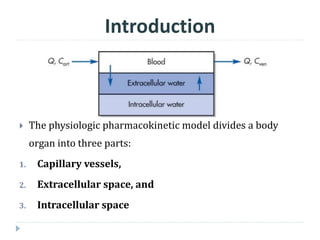
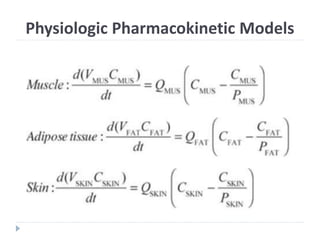
The document discusses physiologic pharmacokinetic models that describe drug movement and disposition in the body, emphasizing factors like organ blood flow and tissue/blood partition coefficients. It outlines the difference between blood flow-limited and diffusion-limited models, detailing how drug concentration changes and elimination processes occur in the body. Additionally, it introduces the concept of mean residence time (MRT), explaining its significance in understanding how long drugs stay in the body after administration.