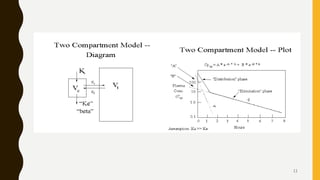
This document discusses pharmacokinetic models, including compartment models. It begins by defining pharmacokinetics and describing different types of pharmacokinetic models. It then focuses on compartment models, explaining that the body can be divided into compartments that exchange materials. It describes multi-compartment models and the two-compartment open model in particular. For the two-compartment model, it outlines the parameters such as apparent volume of distribution, elimination rate constant, and biological half-life. It also discusses nonlinear pharmacokinetics and the Michaelis-Menten equation for describing nonlinear processes.




























![Nonlinear pharmacokinetics.pptx1[1]](https://cdn.slidesharecdn.com/ss_thumbnails/nonlinearpharmacokinetics-201215071246-thumbnail.jpg?width=640&height=640&fit=bounds)



























































