Downloaded 53 times


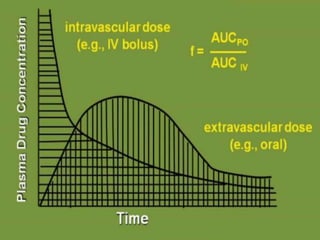


![• E.g.:
Drug products Dose (mg) AUC (mcg/ml/hr)
Oral tablet (a) 200 mg 89.5
Oral suspension(b) 200 mg 86.1
I.V. Bolus 50 mg 37.8
Relative bioavailability = [AUC]a = 89.5 = 1.04
[AUC]b 86.1
= 1.04 X 100 = 104 %
Absolute bioavailability = [AUC] po /Dose PO
[AUC]iv / Dose IV
=89.5/200 = .59
37.8/50
= 59%
Clearance = Dose = 200 = 2.234ml/min/kg
AUC 89.5](https://image.slidesharecdn.com/pkineticparameters-150113093759-conversion-gate01/85/P-kinetic-parameters-8-320.jpg)
![Factors affecting Bioavailability of a Drug
Physical properties of a drug
Physical state:
• Liquids > Solids
[ Solution > Suspension > Capsule > Tablet > Coated tablet ]
Crystalloids > Colloids
Lipid or water solubility:
• Aqueous phase at absorption site
• Passage across Cell surface
Dosage forms
Particle size:
• Important for sparingly soluble drugs
• ↓ the size, ↑ the absorption, ↓ the dose
• Nano-crystalline formulations of Saquinavir
• If ↓ absorption needed (local action on GIT), ↑ the size](https://image.slidesharecdn.com/pkineticparameters-150113093759-conversion-gate01/85/P-kinetic-parameters-9-320.jpg)



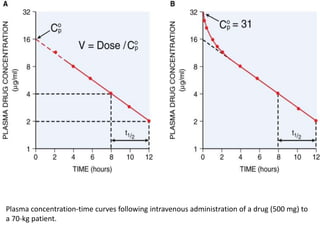

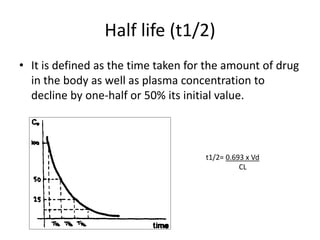
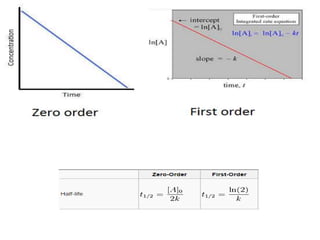


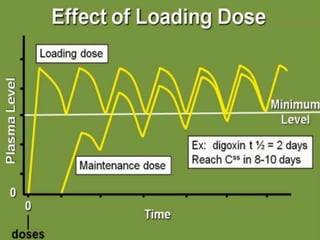

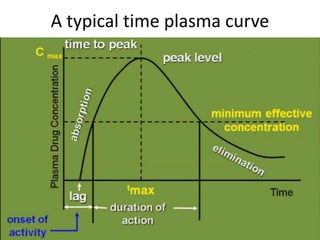
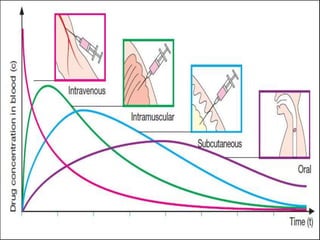
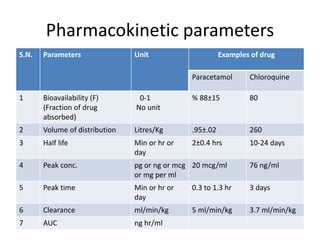
Bioavailability refers to the percentage of an administered drug dose that reaches systemic circulation in an unchanged form. It is calculated as the bioavailable dose divided by the administered dose. Absolute bioavailability compares bioavailability of a non-intravenous dose to an intravenous dose, while relative bioavailability compares bioavailability between different formulations of the same drug. Many factors can affect a drug's bioavailability including its physical properties, the dosage form, physiological factors like pH and transit time, and first-pass metabolism. Volume of distribution represents the hypothetical volume that the drug distributes into in the body and half-life is the time for a drug amount or concentration to reduce by half, which is affected by volume of distribution and clearance.















































