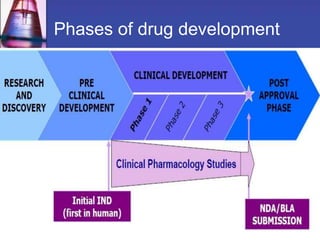
Phase 1 clinical trials are the first studies done in humans of a new drug or treatment. They aim to determine the drug's safety and side effects, identify the maximum tolerated dose, and understand how the body processes the drug through pharmacokinetic evaluation. Phase 1 trials typically involve small groups of healthy volunteers or patients and start with low doses that are gradually increased. The results of phase 1 trials provide information needed to design subsequent clinical trial phases that further evaluate efficacy.










![Method of First In Man study
Done in small number of healthy volunteers or in certain cases
patients
First in a small group of 20 to 25
Start with a dose adjusted from animal data
Slowly increase the dose to find a safe tolerated dose
If safe in a larger group of up to about 50 –75
Randomized , placebo controlled studies.
Performed by clinical pharmacologists
Performed after receiving ethics committee clearance and proper
regulatory approval.
Centre has emergency care & facility for kinetics study
Performed in a single centre
Takes 3 – 6 months [ 70% success rate]](https://image.slidesharecdn.com/phaseclinicaltrial-190213105041/85/Phase-clinicaltrial-12-320.jpg)



![Execution of phase 1-Subject
selection
Inclusion criteria
Healthy volunteers ,
Uniformity of subjects
about age, sex,
nutritional status
[Informed consent a
must]
Exception:
Patients only for toxic
drugs Eg AntiHIV,
Anticancer
Exclusion criteria
Women of child bearing
age, children](https://image.slidesharecdn.com/phaseclinicaltrial-190213105041/85/Phase-clinicaltrial-16-320.jpg)





















































































