Downloaded 262 times


![Introduction
• Since Mizoroki[1] developed the first palladium catalysed reaction, research in this area has
developed exponentially, with each new issue of Angewandte Chemie or JACS highlighting the
latest techniques and processes.
• These reactions show a breadth of applications, not just in the type of transformation, but in the
target structure and scale of the process. Indeed, it is common to see the retrosynthesis of
industrial targets hinge upon a crucial palladium-mediated reaction.
Pd
1. T. Mizoroki, K. Mori, A. Ozaki, Bull. Chem. Soc. Jpn. 1971, 44, 581
(There is still some debate as to which coupling was developed first; many claim that the Kumada coupling of sp2 grignard reagents with
aryl, vinyl or alkyl halides was the first. However, the intrinsic reactivity of grignard reagents with other common functionalities mean that
this coupling is seldom used.)](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-2-320.jpg)
![Why Palladium?
• Why is palladium such an adept catalyst centre? Why not sodium?
• The reason seems to be based around its electronegativity, which leads to relatively strong Pd-H
and Pd-C bonds, and also develops a polarised Pd-X bond.
• It allows easy access to the Pd (II) and Pd (0) oxidation states, essential for processes such as
oxidative addition, transmetalation and reductive elimination,
• Pd (I), Pd (III) and Pd (IV)[2] complexes are also known, though less thoroughly, with Pd (IV)
species essential in C-H activation mechanisms.
2. Pd (VI) complexes has also been proposed (W. Chen, S. Shimada, M. Tanaka, Science, 2002, 295, 308), but theoretical articles
counter-argue this (E. C. Sherer, C. R. Kinsinger, B. L. Kormos, J.D. Thompson, C. J. Cramer Angew. Chem., Int. Ed. 2002, 41, 1953).
The debate is ongoing.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-3-320.jpg)
![The Heck Reaction
• Broadly defined as the palladium-catalyzed coupling of alkenyl or aryl
(sp2) halides or triflates with alkenes to yield products which formally
result from the substitution of a hydrogen atom in the alkene coupling
partner.
• First discovered by Mizoroki, though developed and applied more
thoroughly by Richard F. Heck in the early 1970s.[3]
• Generally thought of as the original palladium catalysed cross-coupling,
and probably the best evolved, including a multitude of
asymmetric varients.[4]
H
R1
R2
R3
cat. [Pd0Ln]
R4 X R4
R1
R2
R3
base
R4 = aryl, benzyl, vinyl
X = Cl, Br, I, OTf
3. R. F. Heck, J. P. Nolley, Jr., J. O rg . Che m . 1972, 3 7 , 2320
4. Review on asymmetric Heck reactions: A. B. Dounay, L. E. Overman, Che m . Re v. 2003, 1 0 3 , 2945 – 2963](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-4-320.jpg)

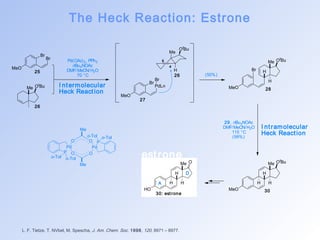
![The Heck Reaction: Taxol
OTf
O
O
O
Me
OTBS
Me
H
BnO
O
[Pd(PPh3)4] (110 mol%)
I nt ramolecular
Heck React ion
O
O
O
M. S. (4 A)
K2CO3, MeCN, 90 °C
(49%)
Me
OTBS
Me
H
BnO
O
22
AcO
O
HO
BzO
Me
OH
Me
H
AcO
O
O
O
taxol
BzHN
Ph
OH
23
24: t axol
a) S. J. Danishefsky, J. J. Masters, W. B. Young, J. T. Link, L. B. Snyder, T. V. Magee, D. K. Jung, R. C. A. Isaacs, W. G. Bornmann, C. A.
Alaimo, C. A. Coburn, M. J. Di Grandi, J. Am. Chem. Soc. 1996, 118, 2843 – 2859
b) J. J. Masters, J. T. Link, L. B. Snyder, W. B. Young, S. J. Danishefsky, Angew. Chem. Int. Ed. Engl. 1995, 34, 1723 – 1726.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-10-320.jpg)
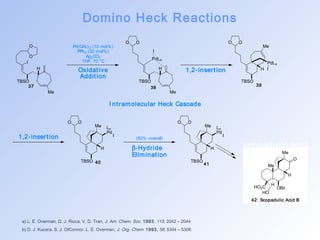
![Domino Heck Reactions
Me
EtO2C
EtO2C
I
Me
EtO2C
EtO2C
I
Y. Zhang, G.Wu, G. Angel, E. Negishi, J. Am. Chem. Soc. 1990, 112, 8590 – 8592.
Me
EtO2C
EtO2C
[Pd(PPh3)4] (3 mol%)
Et3N (2 eq.)
MeCN, 85 °C
(76%)
I nt ramolecular
Domino Heck
32 Cyclisat ion 33](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-12-320.jpg)
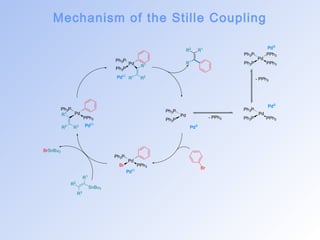
![The Stille Coupling
• Originally discovered by Kosugi et al[5] in the late 1970s, the Stille Coupling was later developed
as a tool for organic transformations by the late Professor J. K. Stille.[6]
• Milder than the older Heck reaction, and more functional-group tolerant, the Stille coupling
remains popular in organic synthesis.
R1 R2 X cat. [Pd0Ln]
SnR3 R1 R3
base
R1 = alkyl, alkynyl, aryl, vinyl
R2 = acyl, alkynyl, allyl, aryl, benzyl, vinyl
X = Br, Cl, I, OAc, OP(=O)(OR)2, OTf
5. Original Report; a) M. Kosugi, K. Sasazawa, Y. Shimizu, T. Migita, Chem. Lett. 1977, 301 – 302; b) M. Kosugi, K. Sasazawa, T. Migita,
Chem. Lett. 1977, 1423 – 1424.
• 6. A a) close D. Milstein, relative J. K. Stille, of the J. Am. Stille Chem. coupling Soc. 1978, is 100the , 3636 Hiyama; – 3638; b) this D. Milstein, involves J. K. the Stille, palladium J. Am. Chem. catalysed Soc. 1979, 101reaction
, 4992 –
4998; c) For a review of Stille Reactions, see; V. Farina, V. Krishnamurthy,W. J. Scott, Org. React. 1997, 50, 1 – 652
of a organosilicon with organic halides/triflates et c., but requires activation with fluoride (TBAF)
or hydroxide.[7]
7. T. Hiyama, Y. Hatanaka, Pure Appl. Chem. 1994, 66, 1471
8. T. R. Kelly, Tetrahedron Lett. 1990, 31, 161
• It is possible to couple bis-aryl halides using R3Sn-SnR3, in a varient known as a Stille-Kelly
reaction, but the toxicity of these species is a somewhat limiting factor.[8]](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-14-320.jpg)
![The Stille Coupling: Rapamycin
O
Me
O
O N
Me
I
I
O
Me
O
H
O
O
H
OH
Bu3Snn
Me
O
Me
O
Me
72 74
O
"St it ching Cyclisat ion"
a) K. C. Nicolaou, T. K. Chakraborty, A. D. Piscopio, N. Minowa, P. Bertinato, J. Am. Chem. Soc. 1993, 115, 4419 – 4420; K. C. Nicolaou, A. D.
Piscopio, P. Bertinato, T. K. Chakraborty, , N. Minowa, K. Koide, Chem. Eur. J. 1995, 1, 318 –333.
b) A. B. Smith III, S. M. Condon, J. A. McCauley, J. L. Leazer, Jr.,J. W. Leahy, R. E. Maleczka, Jr., J. Am. Chem. Soc. 1995, 117, 5407 – 5408.
Me
Me
OH
OMe Me
Me
H OH
OMe
OMe
SnnBu3
[PdCl2(MeCN)2]
(20 mol%)
iPr2NEt, DMF,
THF, 25°C
I ntermolecular
St ille Coupling
O
O
O N
I
O
Me
O
O
H
OH
H
Me
Me
OH
OMe Me
Me
H OH
OMe
OMe
SnnBu3
I nt ramolecular
St ille Coupling
O
O
O N
Me
O
Me
O
O
H
OH
H
Me
Me
OH
OMe Me
Me
H OH
OMe
OMe
O
O
O N
Me
O
Me
O
O
H
OTIPS
H
Me
Me
OTBS
OMe Me
Me
H TESO
Me
OMe
OMe
SnnBu3
I
1. [PdCl2(MeCN)2] (20 mol%)
iPr2NEt, DMF, THF, 25°C (74%)
2. Deprotection (61%)
I nt ramolecular
St ille Coupling
27%
Overall
rapamycin
75 76: Rapamycin](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-16-320.jpg)
![The Stille Coupling: Dynamycin
TeocN
I
I
O
OH
OH
Me
H
OTBS
Me3Sn SnMe3
[Pd(PPh3)4] (5 mol%)
DMF, 75 °C
81%
Tandem
I nt ermolecular
St ille Coupling
TeocN
O
OH
OH
Me
H
OTBS
dynemicin
HN
O
CO2H
OMe
Me
H
OH
O
O
OH
OH
77 81: (± ) Dynamycin
79
Teoc = 2-(trimethylsilyl)ethoxycarbonyl
a) M. D. Shair, T.-Y. Yoon, K. K. Mosny, T. C. Chou, S. J. Danishefsky, J. Am. Chem. Soc. 1996, 118, 9509 – 9525;
b) M. D. Shair, T.-Y. Yoon, S. J. Danishefsky, Angew. Chem. 1995, 107, 1883 – 1885; Angew. Chem. Int. Ed. Engl. 1995, 34, 1721 – 1723;
c) M. D. Shair, T. Yoon, S. J. Danishefsky, J. Org. Chem. 1994, 59, 3755 – 3757.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-17-320.jpg)
![The Stille Coupling: Sanglifehrin
Me
O O
NH
N
O
O
NH
SnnBu3
Me
HN
Me
OH
O Me
O
Me
O
O O
NH
O
HN
O O
NH
O
HN
sanglifehrin
O
86 87
O
a) K. C. Nicolaou, J. Xu, F. Murphy, S. Barluenga, O. Baudoin, H.-X.Wei, D. L. F. Gray, T. Ohshima, Angew. Chem. Int. Ed. 1999, 38, 2447 –
2451;
b) K. C. Nicolaou, F. Murphy, S. Barluenga, T. Ohshima, H. Wei, J. Xu, D. L. F. Gray, O. Baudoin, J. Am. Chem. Soc. 2000, 122, 3830 – 3838.
I
I
[Pd2(dba)3]•CHCl3
AsPh3, iPr2NEt
DMF, 25 °C, 62%
Chemoselect ive
I nt ramolecular
St ille macrocyclisat ion
N
O
NH
OH
O
Me
Me
O
Me
O Me
Me
I
1. [Pd2(dba)3] •CHCl3
AsPh3, iPr2NEt
DMF, 40°C, 45%
2. aq. H2SO4
THF/H2O
(33%)
I nt ermolecular
St ille Coupling
N
O
NH
OH
O
Me
Me
O
Me
O Me
Me Me
NH
O
Me
OH
Me
Me
Me
Me
Me
NH
O
Me
OH
Me
Me
Me
Me 88
87: sanglifehrin A
SnnBu3
23
22](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-18-320.jpg)
![The Stille Coupling: Manzamine A
CO2Me
NBoc
OTBDPS
O
N
Br
TBDPSO
SnnBu3
[Pd(PPh3)4)] (4 mol%)
toluene, 120 °C
I nt ermolecular
St ille Coupling
109
CO2Me
NBoc
OTBDPS
O
N
TBDPSO
N
O
OTBDPS
TBDPSO
N
H Boc E
110
N
O
H
H
OTBDPS
OTBDPS
CO2Me
NBoc
111
endo -int ramolecular
Diels-Alder React ion
(68% Overall)
manzamine
N NH
N
H
A B
C
D
N
H
H
OH
112: Manzamine A
a) S. F. Martin, J. M. Humphrey, A. Ali, M. C. Hillier, J. Am. Chem. Soc. 1999, 121, 866 – 867;
b) J. M. Humphrey, Y. Liao, A. Ali, T. Rein, Y.-L. Wong, H.-J. Chen, A. K. Courtney, S. F. Martin, J. Am. Chem. Soc. 2002, 124, 8584 – 8592.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-19-320.jpg)
![The Carbonylative Stille Coupling:
Jatrophone
O Me
O
Me
82 83
j at rophone
O Me
O
Me O
Me
Me
Me
Me O
Me
Me
Me
Me
A. C. Gyorkos, J. K. Stille, L. S. Hegedus, J. Am. Chem. Soc. 1990, 112, 8465 – 8472.
O Me
Me
Me
Me
[PdCl2(MeCN)2]
LiCl, CO (50 psi)
DMF, 25 °C
I ntermolecular
Carbonylat ive
SnnBu3 St ille Coupling
OTf
SnnBu3
PdLn
Cl
O Me
O
Me
Me
Me
SnnBu3
53% Overall
Cl
PdLn
O
85: (± )-2-epi-jatrophone 84
Carbonyl
I nsert ion](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-20-320.jpg)
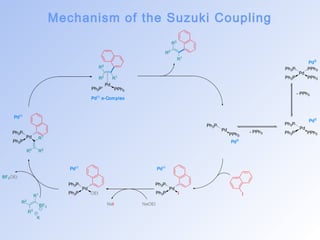
![The Suzuki Coupling
• The Suzuki reaction was formally developed by Suzuki Group in
1979[9], although the inspiration for this work can be traced back
to publications by Heck[10] and Negishi,[11] and their earlier
presentation of these papers at conferences.
• The popularity of this reaction can be partially attributed to the
ease of preparation of the organoboron reagents required, their
general stability, and the lack of toxic by-products.
• Progress in the last quarter-century has shown that the Suzuki
reaction is incredibly powerful, with examples of C(sp2)–C(sp3)
and even C(sp3)–C(sp3) now well documented.[12]
R1 R2 X cat. [Pd0Ln]
BY2 R1 R2
base
R1 = alkyl, alkynyl, aryl, vinyl
R2 = alkyl, alkynyl, aryl, benzyl, vinyl
X = Br, Cl, I, OAc, OP(=O)(OR)2, OTf
9. Original Report; a) N. Miyaura, K. Yamada, A. Suzuki, Tetrahedron Lett. 1979, 20, 3437 – 3440; b) N. Miyaura, A. Suzuki, J. Chem.
Soc. Chem. Commun. 1979, 866 – 867
10. a) R. F. Heck in Proceedings of the Robert A. Welch Foundation Conferences on Chemical Research XVII. Organic-Inorganic Reagents
in Synthetic Chemistry (Ed.W. O. Milligan), 1974, p. 53–98; b) H. A. Dieck, R. F. Heck, J. Org. Chem. 1975, 40, 1083 – 1090.
11. E. Negishi in Aspects of Mechanism and Organometallic Chemistry (Ed.: J. H. Brewster), Plenum, New York, 1978, p. 285.
12. a) T. Ishiyama, S. Abe, N. Miyaura, A. Suzuki, Chem. Lett. 1992, 691 – 694. b) J. Zhou, G.C. Fu, J. Am. Chem. Soc. 2004, 126, 1340 –
1341, and references therein. c) A. C. Frisch, M. Beller, Angew. Chem. Int. Ed. 2005, 44, 674 – 688. d) For a relatively recent review,
see N. Miyaura, A. Suzuki, Chem. Rev. 1995, 95, 2457.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-21-320.jpg)
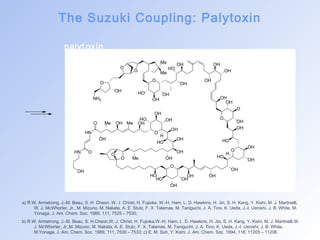
![The Suzuki Coupling: Palytoxin
O
O
OTBS
NHTeoc
Me
O Me
O
TBSO OTBS
TBSO OTBS
OTBS
B
OTBS
TBSO
TBSO
OTBS
HO
OH
O
OAc I
OTBS
TBSO OTBS
OTBS
OTBS
O OTBS
CO2Me
TBSO
TBSO
H
OTBS
I ntermolecular
Suzuki Coupling
[Pd(PPh3)4] (40 mol%)
TlOH, THF/H2O, 25 °C
(70%)
O
O
OTBS
TeocHN
O
Me
Me
O
TBSO
TBSO
OTBS
TBSO OTBS OTBS
OTBS
TBSO OTBS
O
OAc
OTBS
OTBS
OTBS
OTBS
TBSO
O
MeO2C
TBSO H
OTBS
OTBS OTBS
a) R.W. Armstrong, J.-M. Beau, S. H. Cheon, W. J. Christ, H. Fujioka, W.-H. Ham, L. D. Hawkins, H. Jin, S. H. Kang, Y. Kishi, M. J. Martinelli,
W. J. McWhorter, Jr., M. Mizuno, M. Nakata, A. E. Stutz, F. X. Talamas, M. Taniguchi, J. A. Tino, K. Ueda, J.-I. Uenishi, J. B. White, M.
Yonaga, J. Am. Chem. Soc. 1989, 111, 7525 – 7530;
b) R.W. Armstrong, J.-M. Beau, S. H.Cheon,W. J. Christ, H. Fujioka,W.-H. Ham, L. D. Hawkins, H. Jin, S. H. Kang, Y. Kishi, M. J. Martinelli,W.
J. McWhorter, Jr.,M. Mizuno, M. Nakata, A. E. Stutz, F. X. Talamas, M. Taniguchi, J. A. Tino, K. Ueda, J.-I. Uenishi, J. B. White,
M.Yonaga, J. Am. Chem. Soc. 1989, 111, 7530 – 7533;
c) E. M. Suh, Y. Kishi, J. Am. Chem. Soc. 1994, 116, 11205 – 11206.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-23-320.jpg)
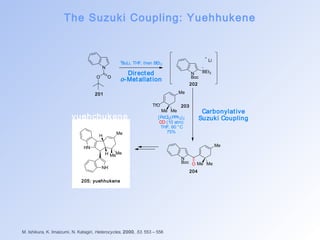
![The Suzuki Coupling: FR182887
MeO
O
Me
Br
OTBS
Me Me Br
126 127
O
OTBS
HO
H Me
HO
[PdCl2(dppf))] (10 mol%)
Cs2CO3, DMF/H2O, 100 °C
O Me
B
HO
H Me
HO
HO
H Me
O
O
130 129
a) D. A. Evans, J. T. Starr, J. Am. Chem. Soc. 2003, 125, 13531 –13540
b) D. A. Evans, J. T. Starr, Angew. Chem. 2002, 114, 1865 – 1868; Angew. Chem. Int. Ed. 2002, 41, 1787 – 1790.
TBDPSO
B
OTBS
Me
OTBS
HO
OH
[Pd(PPh3)4)] (5 mol%)
Tl2CO3, THF/H2O, 23 °C
(84%)
I ntermolecular
Suzuki Coupling
TBDPSO
OTBS
Me
OTBS
MeO
Me
Me Me Br
O
OH
Br
H H
H
CO2Et
H
Me
Me
H
B
O
B
O
Me
Me
(71%)
O
OH
Me
H H
H
CO2Et
H
Me
Me
H O
OH
Me
H H
H
H
Me
Me
H
fr182887
131 132: FR182887
128
I ntermolecular
Suzuki Coupling](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-25-320.jpg)
![The Suzuki Coupling: Dragmacidin Me
TBSO
HO
Br
O N SEM
[Pd(PPh3)4] (10 mol%)
toluene/MeOH/H2O, 23 °C
I ntermolecular
Heck React ion
Me
TBSO
HO
PdOAc
O N SEM
162 164
TBSO
HO
O
(74%)
N SEM
H
TBSO
MeO
H
O
O N SEM
B O
166 165
[Pd(PPh3)4] (10 mol%)
161, toluene/MeOH/H2O
NaCO3, 50 °C, 77%
I ntermolecular
Suzuki React ion
TBSO
MeO
H
O N SEM
NTs
Br
N
N
OMe
167
dragmacidin
HO
Me NH
H
O NH
N
HO N
159 160
N
Br OMe
a) N. K. Garg, D. D. Capsi, B. M. Stoltz, J. Am. Chem. Soc. 2004, 126, 9552 – 9553.
b) For a failed alternative route without Pd Catalysis: N. K. Garg, R. Sarpong, B. M. Stoltz, J. Am. Chem. Soc. 2002, 124, 13179 – 13184.
Br
NH
O
N
N
H2N
168: dragmacidin
Ts
N
B
Br
OH
N I
Br OMe
[Pd(PPh3)4] (10 mol%)
toluene/MeOH/H2O, 23 °C
(71%)
I ntermolecular
Suzuki
Coupling
NTs
Br
N
161](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-26-320.jpg)
![The Suzuki-Miyaura B-Alkyl Coupling: CP-236,114
O
TBS I
169 170
173 171 CP-263,114
13) a) N. Miyaura, T. Ishiyama, M. Ishikawa, A. Suzuki, Tetrahedron Lett. 1986, 27, 6369 – 6372; b) not to be confused with the Miyaura
boration, in which an aryl halide is converted to an aryl boronate via palladium catalysis and a diboron reagent. However, this is a useful
preparation of the organoboron reagents required for the Suzuki reaction. See: T. Ishiyama, M. Murata, N. Miyuara. J. Org. Chem.
1995, 60, 7508.
14) Review of the development, mechanistic background, and applications of the B-alkyl Suzuki-Miyaura cross-coupling reaction, see S. R.
Chemler, D. Trauner, S. J. Danishefsky, Angew. Chem. Int. Ed. 2001, 40, 4544 – 4568.
15) Q. Tan, S. J. Danishefsky, Angew. Chem. Int. Ed. 2000, 39, 4509 – 4511.
O
TBSO
H
O
TBS
O
H
OTBS
O
TBS
OTBS
H
OTBS
I
O
TBS
OTBS
H
OTBS
OBn
6
O
O
O
O O
O
CO2H
H
Me
O
H
Me
[Pd(OAc)2(PPh3)2]
Et3N, THF, 65 °C
(92%)
I nt ermolecular
Heck React ion
B{ (CH2)6OBn} 3
[PdCl2(dppf)]
CsCO3, AsPh3, H2O, 25 °C
(70%)
Suzuki-Miyaura
B-Alkyl React ion
174: CP-263,114
• An important trend in Suzuki
chemistry is the development of a
C(sp3)–C(sp2) methodology, which
has become known as the Suzuki-
Miyaura B-Alkyl varient.[13-15]
• Often used as an alternative to
RCM, leaving a single isolated
double bond, rather than the
conjugated systems produced by a
regular Suzuki coupling.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-27-320.jpg)
![The Suzuki Coupling: Phomactin A
O
OTMS
O
H
Me
Me
OTES
Me
I
9-BBN
THF, 40 °C
phomact in
O
a) P. J. Mohr, R. L. Halcomb, J. Am. Chem. So c. 2003, 125, 1712 – 1713
b) N. C. Callan, R. L. Halcomb, Org. Lett. 2000, 2, 2687 – 2690.
O
Me OTMS
O
H
Me
OTES
Me
I
B
O
O
Me H
OTMS
OTES
Me
Me
Me
O
Me H
OH
OH
Me
Me
Me
TBAF
(78%)
Suzuki-Miyaura
B-Alkyl
Macrocyclisat ion
[PdCl2(dppf)] (100 mol%)
AsPh3(200 mol%), Tl2CO3
THF/DMF/H2O, 25 °C
(37%)
200: phomact in A](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-28-320.jpg)
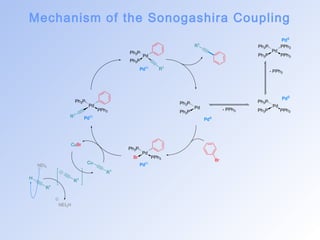
![The Sonogashira Coupling
• The coupling of terminal alkynes with vinyl or aryl halides via palladium catalysis was first
reported independently and simultaneously by the groups of Cassar[16] and Heck[17] in 1975.
• A few months later, Sonogashira and co-workers demonstrated that, in many cases, this cross-coupling
reaction could be accelerated by the addition of cocatalytic CuI salts to the reaction
mixture.[18,19]
• This protocol, which has become known as the Sonogashira reaction, can be viewed as both an
alkyne version of the Heck reaction and an application of palladium catalysis to the venerable
Stephens–Castro reaction (the coupling of vinyl or aryl halides with stoichiometric amounts of
copper(I) acetylides).[20]
• Interestingly, the utility of the “copperfree” Sonogashira protocol (i.e. the original Cassar–Heck
version of this reaction) has subsequently been “rediscovered” independently by a number of
other researchers in recent years.[21]
R2 X
cat. [Pd0Ln]
R1 H R2 R2
16. L. Cassar, J. Organomet. Chem. 1975, 93, 253 – 259.
17. H. A. Dieck, F. R. Heck, J. Organomet. Chem. 1975, 93, 259 – 263.
18. K. Sonogashira, Y. Tohda, N. Hagihara, Tetrahedron Lett. 1975, 16, 4467 – 4470.
19. For a brief historical overview of the development of the Sonogashira reaction, see: K. Sonogashira, J. Organomet. Chem. 2002, 653,
46 – 49.
20. R. D. Stephens, C. E. Castro, J. Org. Chem. 1963, 28, 3313 – 3315.
21. a) M. Alami, F. Ferri, G. Linstrumelle, Tetrahedron Lett. 1993, 34, 6403 – 6406; b) J.-P. Genet, E. Blart, M. Savignac, Synlett 1992, 715
– 717; c) C. Xu, E. Negishi, Tetrahedron Lett. 1999, 40, 431 – 434;
base
R1 = alkyl, aryl, vinyl
R2 = alkyl, benzyl, vinyl
X = Br, Cl, I, OTf](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-30-320.jpg)
![The Sonogashira Coupling: Eicosanoid 212
Br Me
OTBS
TMS
[Pd(PPh3)4] (4 mol%)
CuI (16 mol%)
nPrNH2, C6H6, 25 °C
Sonogashira
Coupling
K. C. Nicolaou, S. E. Webber, J. Am. Chem. Soc. 1984, 106, 5734 – 5736
R
Me
OTBS
AgNO3,
KCN
208: R = TMS
209: R = H
210, [Pd(PPh3)4] (4 mol%)
CuI (16 mol%)
nPrNH2, C6H6, 25 °C
76% Overall from 208
Br
CO2Me
OTBS
Me
OTBS
CO2Me
OTBS
Me
OH
CO2H
OH
Sonogashira
Coupling
206
207
210
212 211](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-32-320.jpg)
![The Sonogashira Coupling: Disorazole C1
PMBO
OH
PMBO
OH
218
[Pd(PPh3)2Cl2] (4 mol%)
CuI (30 mol%), Et3N
MeCN, -20 °C, 94%
217 219
O
O OMe
N
N
O
Me Me
P. Wipf, T. H. Graham, J. Am. Chem. Soc. 2004, 126, 15346 –15347.
Me
Me
Me
Me
Me
Me
N
CO2Me
MeO O
Sonogashira
Coupling
220, DCC, DMAP
80%
PMBO
Me
O
Me
Me
OMe
N
CO2Me
N
O
MeO O
O
I
218
[Pd(PPh3)2Cl2] (5 mol%)
CuI (20 mol%), Et3N
MeCN, -20 °C, 94%
Sonogashira
Coupling
PMBO
Me
O
Me
Me
O OMe
N
Me Me
CO2Me
N
O
MeO O
OH
Me
OPMB
Me
OH
Me
Me
MeO O
O
OH
Me
O
disorazole
N
O
RO
O
I
OMe
218: R = Me
220: R = H
221
223: Disorazole C1 222](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-33-320.jpg)
![The Sonogashira Coupling: Dynemicin
MeO2CN
OMe
Me
O
O
Br
MeO2CN
OMe
Me
O
[Pd(PPh3)4] (2 mol%)
CuI (20 mol%)
toluene, 25 °C
I nt ramolecular O
Sonogashira
Coupling
243 244
MeO2CN
OMe
Me
O
O
H
H
244
H
H
MeO2CN
OMe
Me
OH
246
Br
1) CO2Me
[Pd(PPh3)4] (2 mol %)
CuI (20 mol %)
toluene, 25 °C
2) LiOH, THF/H2O
65% overall
Sonogashira
Coupling
MeO2CN
2,4,6-Cl3C2H2COCl
DMAP, toluene, 25 °C
OMe
Me
Diels-
Alder
CO2H
OH
50%
248
247
Yamaguchi
Macrolactonisat ion/
Diels-Alder
HN
OMe
Me
H
O
O
O
OMe
OMe
CO2Me
OMe
dynemicin
249: t ri-O- methyl dynemicin A
met hyl est er
a) J. Taunton, J. L. Wood, S. L. Schreiber, J. Am. Chem. Soc. 1993, 115, 10 378 – 10379
b) J. L. Wood, J. A. Porco, Jr., J. Taunton, A. Y. Lee, J. Clardy, S. L. Schreiber, J. Am. Chem. Soc.
1992, 114, 5898 – 5900
c) H. Chikashita, J. A. Porco, Jr., T. J. Stout, J. Clardy, S. L. Schreiber, J. Org. Chem. 1991, 56, 1692 – 1694
d) J. A. Porco, Jr., F. J. Schoenen, T. J. Stout, J. Clardy, S. L. Schreiber, J. Am. Chem. Soc. 1990, 112, 7410 – 7411.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-34-320.jpg)
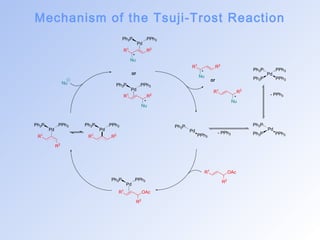
![The Tsuji-Trost Reaction
• The palladium catalysed nucleophilic substitution of allylic
compounds was discovered independently by Trost and Tsuji, and
represents the first example of a metalated species acting as an
electrophile.[22]
• Originally developed as a stoichiometric process, Trost succeeded in
transforming the allylation of enolates with p-allyl–palladium
complexes into the catalytic process of renown.[23,24]
• A wide range of allylic substrates undergo this reaction with a
correspondingly wide range of carbanions, making this a versatile
and important process for the formation of carbon–carbon bonds.
• Whilst the most commonly employed substrates for palladium-catalyzed
allylic alkylation are allylic acetates, a variety of leaving
groups also function effectively—these include halides, sulfonates,
carbonates, carbamates, epoxides, and phosphates.
cat. [Pd0Ln]
X NuH Nu
base
X = Br, Cl, OCOR, OCO2R, CO2R, P(=O)(OR)2
NuH = b-dicarbonyls, b-ketosulfones, enamines, enolates
22. For early reviews of the Tsuji-Trost reaction, see a) B. M. Trost, Acc. Chem. Res. 1980, 13, 385 – 393; b) J. Tsuji, Tetrahedron 1986,
42, 4361 – 4401.
23. J. Tsuji, H. Takahashi, Tetrahedron Lett. 1965, 6, 4387 – 4388.
24. For recent reviews of the palladium-catalyzed asymmetric alkylation reaction, see: a) B. M. Trost, M. L. Crawley, Chem. Rev. 2003,
103, 2921 – 2943; b) B. M. Trost, J. Org. Chem. 2004, 69, 5813 – 5837.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-35-320.jpg)
![The Tsuji-Trost Reaction: Strychnine
O
PdL AcO O OMe n
O
tBuO CO2Et
[Pd2(dba)3] (1 mol%)
PPh3 (15 mol%)
NaH, THF, 23 °C
[-CO2, -MeO ]
Tsuj i-Trost
React ion
AcO
O
tBuO CO2Et
Me
N
O
st rychnine
H
H
H
H
a) S. D. Knight, L. E. Overman, G. Pairaudeau, J. Am. Chem. Soc. 1993, 115, 9293 – 9294
b) S. D. Knight, L. E. Overman, G. Pairaudeau, J. Am. Chem. Soc. 1995, 117, 5776 – 5788.
AcO
OtBu
O
H
CO2Et
91%
Me3Sn
TIPSO
OtBu
[Pd2(dba)3] (3 mol%)
AsPh3 (22 mol%), CO (50 psi)
LiCl, NMP, 70 °C
80%
Carbonylat ive
St ille Coupling
TIPSO
OtBu
O
N
MeN
N
O O
250
251
252
253
MeN
N
NMe
O
I
256: St rychnine 255 254](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-37-320.jpg)
![OTBS
O
PhO2S
MeO2C
The Tsuji-Trost Reaction: Roseophilin
[Pd2(dba)3] (1 mol%)
PPh3 (15 mol%)
NaH, THF, 23 °C
Tsuj i-Trost
Macrocyclisat ion
LnPd O
TBSO
PhO2S
MeO2C
LnPd OH
TBSO
PhO2S
MeO2C
263 264 265
PhO2S PhO2S
O O
O
MeO2C HO
OTBS
-[Pd0Ln]
85%
BnNH2
[Pd(PPh3)4] (15 %)
THF, 35 °C, 70%
Tsuj i-Trost
O React ion
PhO2S
NBn
HO
268 267 266
Roseophilin
N
Me
O
Me
MeO
Cl NH
269: Roseophilin
a) A. Fürstner, H. Weintritt, J. Am. Chem. Soc. 1998, 120, 2817 – 2825;
b) A. Fürstner, T. Gastner, H. Weintritt, J. Org. Chem. 1999, 64, 2361 – 2366.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-38-320.jpg)
![The Tsuji-Trost Reaction: Hamigeran B
Pd
Me
P P
b
Me
B. M. Trost, C. Pissot-Soldermann, I. Chen, G.M. Schroeder, J. Am. Chem. Soc. 2004, 126, 4480 – 4481.
O
OtBu
OAc
[{h3-C3H5PdCl} 2] (1 mol%)
ligand 285 (2 mol%)
LDA, tBuOH, Me3SnCl
DME, 25 °C
O
Asymmet ric
Allylic Alkylat ion Me
tBuO
P P
Pd
a
*
*
O
OtBu
77%, 93% ee
OMe O
Me OTf
Me
Me Me
Pd(OAc) (10 mol%)
dppb (20 mol%)
K2CO3
toluene, 110 °C, 58%
I nt ramolecular
Heck React ion
OMe O
Me H
Me
Me
Me
OMe O
Me H
Me
Me
Me
NH
O
P
Ph
Ph
HN
O
P
Ph
Ph
hamigeran B
285
284
286
287
288
290: hamigeran 289](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-39-320.jpg)
![The Tsuji-Trost Reaction: (+)-g-lycorane
* 66%, 54% ee
OBz
BzO OBz
NH
291
MeO2C
O
O
O Br
[Pd2(OAc)3] (5 mol%)
293 (10 mol%)
LDA
THF/MeCN, 0 °C
Asymmet ric
Allylic Alkylat ion
O
O
Br
NH
O
MeO2C
P P
Pd
294 295
O PdLn
O
H
H
H
O H
lycorane
O
H H H
H. Yoshizaki, H. Satoh, Y. Sato, S. Nukui, M. Shibasaki, M. Mori, J. Org. Chem. 1995, 60, 2016 – 2021.
O
O
Br
NH
O
MeO2C
OBz
Pd(OAc) (5 mol%)
dppb (20 mol%)
NaH
DMF, 50 °C
I nt ramolecular
Allylic Alkylat ion/
Heck React ion
Cascade
O
O
Br
N
MeO2C
O O
Br
N
MeO2C
H
iPr2NEt, 100 °C
O O
N
CO2Me
O
N
299: (+ ) -g-lycorane
298
297 296
292
O
O
PPh2
PPh2
293](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-40-320.jpg)
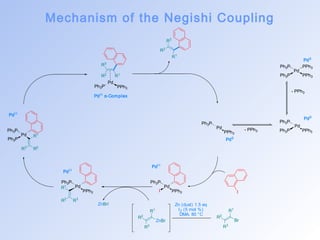
![The Negishi Coupling
• The use of organozinc reagents as the nucleophilic component in
palladium-catalyzed cross-coupling reactions, known as the Negishi
coupling, actually predates both the Stille and Suzuki processes,
with the first examples published in the 1970s.[25]
• However, the stunning progress in the latter procedures left the
Negishi process behind, underappreciated and underutilised.
• Organozinc reagents exhibit a very high intrinsic reactivity in
palladium-catalyzed cross-coupling reactions, which combined with
the availability of a number of procedures for their preparation and
their relatively low toxicity, makes the Negishi coupling an
exceedingly useful alternative to other cross-coupling procedures, as
well as constituting an important method for carbon–carbon bond
formation in its own right.[26]
ZnR2 R1 R3
25. a) E. Negishi, A. O. King, N. Okukado, J. Org. Chem. 1977, 42, 1821 – 1823; for a discussion, see: b) E. Negishi, Acc. Chem. Res.
1982, 15, 340 – 348.
26. a) E. Erdik, Tetrahedron 1992, 48, 9577 – 9648; b) E. Negishi, T. Takahashi, S. Babu,D. E. Van Horn, N. Okukado, J. Am. Chem. Soc.
1987, 109, 2393 – 2401.
R1 R3 X
cat. [Pd0Ln]
R1 = alkyl, alkynyl, aryl, vinyl
R3 = acyl, aryl, benzyl, vinyl
X = Br, I, OTf, OTs](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-41-320.jpg)
![The Negishi Coupling: Discodermolide
Me
Me
Me
Me
309 310 312
Me
discodermolide
Me
O O
O
HO
a) A. B. Smith III, T. J. Beauchamp, M. J. LaMarche, M. D. Kaufman, Y. Qiu, H. Arimoto, D. R. Jones, K. Kobayashi, J. Am. Chem. Soc. 2000,
122, 8654 – 8664;
b) A. B. Smith III, M. D. Kaufman, T. J. Beauchamp,M. J. LaMarche, H. Arimoto, Org. Lett. 1999, 1, 1823 – 1826.
c) For a review of the chemistry and biology of discodermolide, see: M. Kalesse, ChemBioChem 2000, 1, 171 – 175
d) For examples of other approaches to discodermolide, see: I. Paterson, G. J. Florence, Eur. J. Org. Chem. 2003, 2193 – 2208.
e) In the synthesis of discodermolide by the Marshall group, a B-alkyl Suzuki–Miyarua fragment-coupling strategy was employed to form the
C14C15 bond, in which 2.2 equivalents of an alkyl iodide structurally related to 309 was required: J. A. Marshall, B. A. Johns, J. Org.
Chem. 1998, 63, 7885 – 7892.
I
Me Me
TBSO O O
PMP
tBuLi, ZnCl2
Et2O
-78 °C Zn
Me Me
TBSO O O
PMP
[Pd(PPh3)4] (5 mol%)
311
Et2O, 25 °C, 66%
Negishi Coupling
Me Me
OTBS O O
PMP
Me
PMBO
Me
OTBS
I
PMBO
Me
OTBS
Me
= 311
Me Me
OH O
Me
Me Me
OH
NH2
HO
Me
HO
313: discodermolide
15 15
15
14
14
15
14](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-43-320.jpg)
![The Negishi Coupling: Amphidinolide T1
Cl O
Me
O
O
Me
TBDPSO Me
O
Me
OMOM
R
314: R = ZnI
(315: R = I)
(316: R = H)
[Pd2(dba)3] (3 mol%)
285
P(2-furyl)3 (6 mol %)
toluene/DMA, 25 °C, 50%
Negishi Coupling
Me O
amphidinolide
a) C. Aïssa, R. Riveiros, J. Ragot, A. Fürstner, J. Am. Chem. Soc. 2003, 125, 15 512 – 15520.
OMOM
TBDPSO Me
Me
O
O
Me
O
Me O
OMOM
TBDPSO Me
O
Me Me
O
O
317
318
319: Amphidinolide T1](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-44-320.jpg)
![The Fukuyama Coupling
• The Fukuyama Coupling is a
modification of the Negishi
Coupling, in which the electrophilic
component is a thioester.
• The product of the coupling with a
Negishi-type organozinc reagent is
carbonyl compound, thus negating
the need for a carbon monoxide
atmosphere.
O
SR4
R1 R3 cat. [Pd0Ln]
ZnR2 R1 R3
R1 = alkyl, alkynyl, aryl, vinyl
R3 = acyl, aryl, benzyl, vinyl
R4 = Me, Et, et c.
O ZnI [PdCl2(PPh3)2] (10 mol%)
27) H. Tokuyama, S. Yokoshima, T. Yamashita, S.-C. Lin, L. Li, T. Fukuyama, J. Braz. Chem. Soc., 1998, 9, 381-387.
O
MeO
SEt
toluene, 25 °C, 5 min, 87%
Fukuyama Coupling MeO
O](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-45-320.jpg)
![Palladium Catalysis: Outlook And Summary
• This review has highlighted only a small number of applications of palladium catalysis in organic
synthesis, but new examples are published every month.
• Each example pushes the field forwards, towards universal conditions, where application of them
results in a useful yield without prior optimisation.
• However, palladium is only one metal; the breadth of catalysis available from rhodium,[28]
ruthenium[29] and platinum based systems extend far further, and into the realms of metathesis.[30]
Fürstner has shown analogous procedures using Iron catalysts,[31] with obvious economic and
toxicity benefits.
28) For an example of palladium-mimicking rhodium catalysis, see: M. Lautens and J. Mancuso, Org. Lett. 2002, 4, 2105
29) For a recent review of "atom ecconomic" ruthenium catalysis, see: B. M. Trost, M. U. Frederiksen, M. T. Rudd, Angew. Chem. Int. Ed.,
2005, 41, 6630 – 6666.
30) For the complementary review on Metathesis Reactions in Total Synthesis, see: K. C. Nicolaou, P. G. Bulger, D. Sarlah , Angew. Chem. Int.
Ed., 2005, 41, 4490-4527.
31) A. Fürstner, R. Martin, Chem. Lett. 2005, 34, 624-629.](https://image.slidesharecdn.com/palladiumcatalysedreactionsinsynthesis-141014223547-conversion-gate02/85/Palladium-catalysed-reactions-in-synthesis-46-320.jpg)

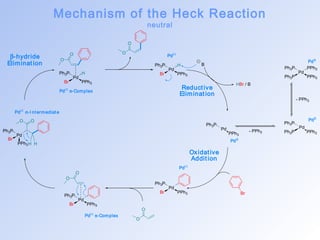
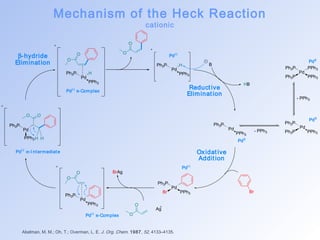
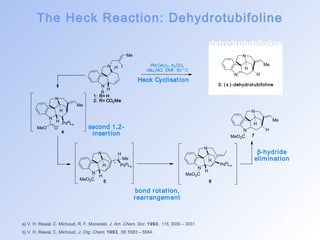
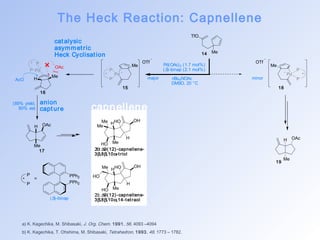
This document summarizes key aspects of palladium-catalyzed cross-coupling reactions, with a focus on the Heck reaction and its mechanisms and applications. The Heck reaction involves the coupling of alkenyl or aryl halides with alkenes, catalyzed by palladium. The mechanism proceeds through oxidative addition, transmetalation, and reductive elimination steps. The document discusses factors that determine regioselectivity and provides examples of the Heck reaction in total syntheses of natural products like dehydrotubifoline, capnellene, and taxol. It also describes domino and intramolecular Heck reactions and summarizes the related Stille coupling reaction.



































































