

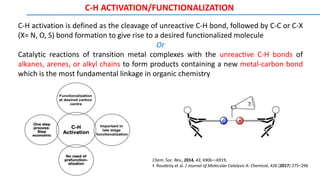

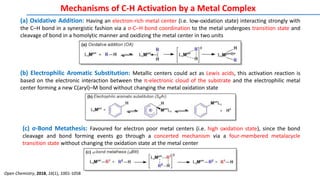
![Different Types of Additives and their Roles in C–H Functionalisation Reactions
Oxidants: Most often Cu salts (commonly Cu(OAc)2), and Ag salts (AgOAc, AgOTf, AgOPiv) sometimes Mn salts,
are used in stoichiometric or superstoichiometric amounts in oxidative reactions. Other oxidants, used alone or in
combination with Cu or Ag are benzoquinones, peroxides, O2/air, K2S2O8 or hypervalent iodine compounds
Catalytic Ag salts: Catalytic amounts of Ag salts are often used in combination with groups 8 or 9 metal halide
dimers, commonly used as catalysts (e.g. [RhCp*Cl2]2). In these cases the Ag acts as a halide scavenger, and the
counteranion (usually OTf, NTf2, or SbF6) promotes the in situ formation of cationic metal catalysts in solution
Carboxylates: Main role of carboxylates is to deprotonate the desired C-H bond, which is to be activated. It occurs
via concerted metalation deprotonation (CMD) mechanism. eg. Cu, Ag, Zn, Na salts of acetates, benzoates,
pivalates, admantanecarboxylates, trifluoroacetates, etc.
Chem. Soc. Rev., 2018, 47, 6603-6743](https://image.slidesharecdn.com/c-hactivationandfunctionalization-191015055756/85/C-H-Activation-and-Functionalization-10-320.jpg)

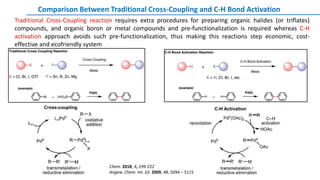
![Palladium(II) Acetate
Palladium(II) Acetate Trimer
Palladium(II) Trifluoroacetate Palladium(II) Acetylacetonate
Bis(acetonitrile)palladium(II)
Dichloride
Bis(tricyclohexylphosphine)-
palladium(II) DichlorideDichloro[9,9-dimethyl-4,5-
bis(diphenylphosphino)xanthene]-
palladium(II)
Tetrakis(triphenylphosphine)-
palladium(0)
Tris(dibenzylideneacetone)-
dipalladium(0)
Bis(dibenzylideneacetone)-
palladium(0)
Tetrakis(acetonitrile)-
palladium(II) Ditriflate
Pd(II)Pd(0)
1,2-Bis(phenylsulfinyl)ethane
palladium(II) acetate
(White Catalyst)
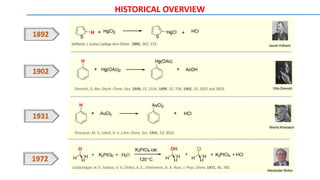
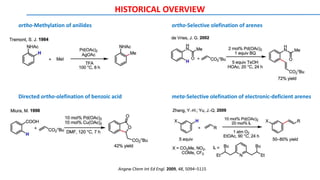
Transition-metal catalysts, especially Pd and Rh catalysts, are crucial for C–H bond cleavage and further
transformations. In general, palladium(II), rhodium(I), iridium(I), ruthenium(II), copper(II), and iron(II) are
widely used in C-H bond activation
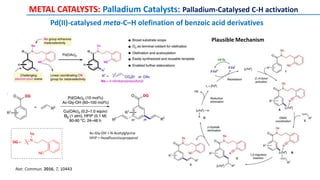
METAL CATALYSTS: Palladium Catalysts](https://image.slidesharecdn.com/c-hactivationandfunctionalization-191015055756/85/C-H-Activation-and-Functionalization-13-320.jpg)
![METAL CATALYSTS: Rhoduim Catalysts
Chlorobis(ethylene)-
rhodium(I) Dimer
Acetylacetonatobis(ethylene)-
rhodium(I)
Chloro(1,5-hexadiene)-
rhodium(I) Dimer
Chloro(1,5-cyclooctadiene)-
rhodium(I) Dimer
Bis(1,5-cyclooctadiene)-
rhodium(I) Tetrafluoroborate
Chlorobis(cyclooctene)-
rhodium(I) Dimer
Norbornadiene
Rhodium(I) Chloride Dimer
Bis[η-(2,5-norbornadiene)]-
rhodium(I) Tetrafluoroborate
(Pentamethylcyclopentadienyl)-
rhodium(III) Dichloride Dimer Tris(triphenylphosphine)rhodium(I)
Chloride
Rhodium(II) Acetate Dimer](https://image.slidesharecdn.com/c-hactivationandfunctionalization-191015055756/85/C-H-Activation-and-Functionalization-17-320.jpg)

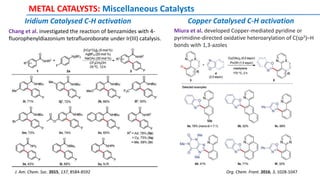
![METAL CATALYSTS: Miscellaneous Catalysts
Iridium
Chlorobis(ethylene)iridium(I)
Dimer
(Acetylacetonato)-
(1,5-cyclooctadiene)iridium(I)
(Pentamethylcyclopentadienyl)-
iridium(III) Dichloride Dimer
Vaska's Catalyst
Copper
Copper(I) Acetate
Copper(I)
2-Thiophenecarboxylate
Tetrakis(acetonitrile)copper(I)
Hexafluorophosphate
Copper(II)
Trifluoromethanesulfonate
Copper(II) Acetylacetonate
Iron
Cyclopentadienyliron Dicarbonyl Dimer
Tris(dibenzoylmethanato) Iron
Iron(II) Acetate
Iron(III) Acetylacetonate
Nickel
Bis(triphenylphosphine)-
nickel(II) Dichloride
[1,3-Bis(diphenylphosphino)-
propane]nickel(II) Dichloride
Nickel(II) Chloride Anhydrous
Nickel(II)
Trifluoromethanesulfonate](https://image.slidesharecdn.com/c-hactivationandfunctionalization-191015055756/85/C-H-Activation-and-Functionalization-19-320.jpg)
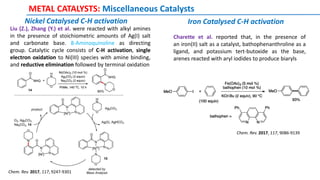

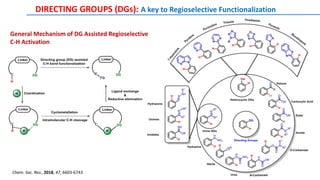
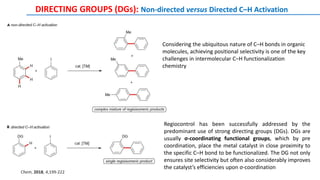
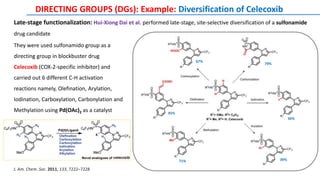


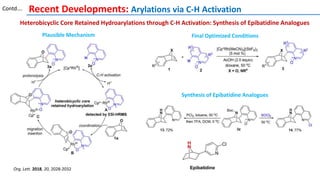
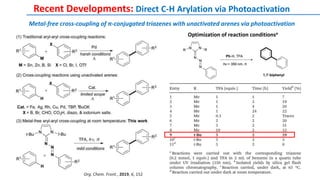


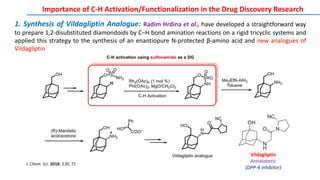
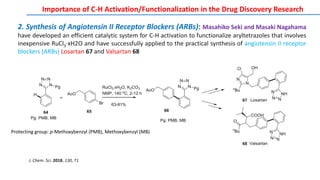
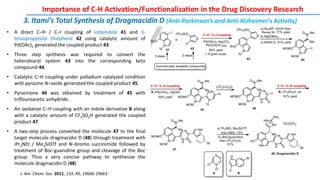



The document discusses the advancements in C-H bond activation and functionalization, highlighting its significance in drug discovery. It contrasts traditional cross-coupling methods with C-H activation approaches, emphasizing the latter's economic and eco-friendly advantages. Additionally, it covers the mechanisms of C-H activation by various metal catalysts, the role of directing groups, and the challenges faced in achieving regioselective functionalization.

























![PHASE TRANSFER CATALYSIS [PTC]](https://cdn.slidesharecdn.com/ss_thumbnails/64-191219161253-thumbnail.jpg?width=640&height=640&fit=bounds)









































