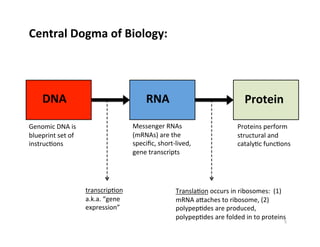
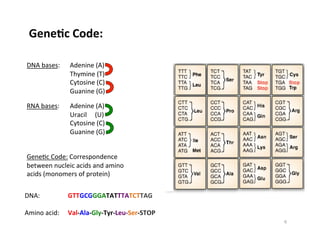
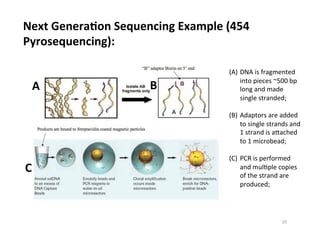
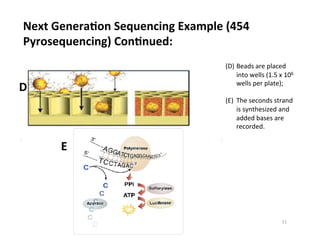
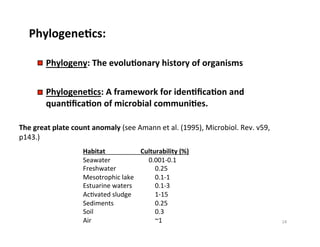
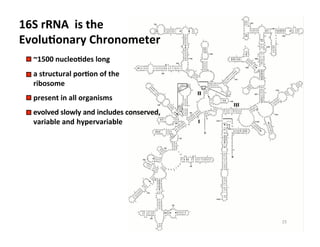
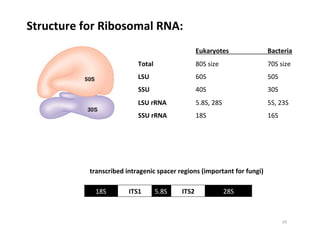
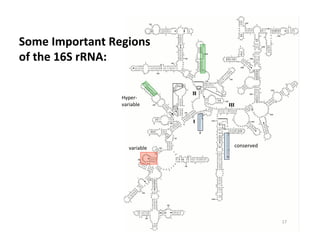
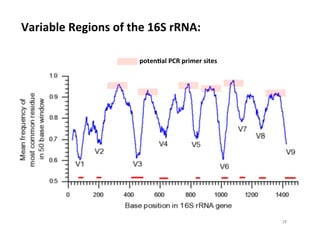
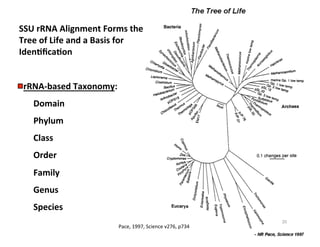
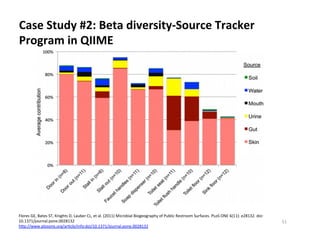
This document outlines molecular biology methods for analyzing bioaerosols, focusing on genetic sequencing techniques such as Sanger and next-generation sequencing. It discusses the central dogma of biology, identifies various molecular techniques for identification and quantification of microorganisms, and addresses challenges in aerosol sample analysis. Additionally, it presents case studies examining microbial diversity in different environments, highlighting the significance of human occupancy as a source of indoor airborne bacteria.













![Reproducibility and RepeatabilityReproducibility and Repeatability
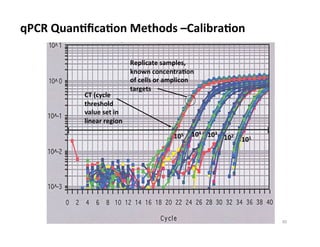
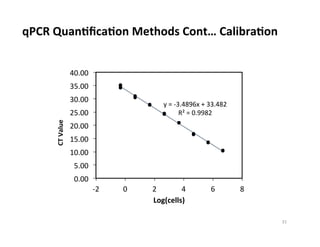
Reproducibility Near Detection Level limit
~103
cells
~104
cells
Copyright
©
American
Society
for
Microbiology,
[doi:
10.1128/AEM.01240-‐10
Appl.
Environ.
Microbiol.
November
2010
vol.
76
no.
21
7004-‐701]
32](https://image.slidesharecdn.com/molecularmethods-bioaerosols-peccia-140324090829-phpapp01/85/DNA-based-methods-for-bioaerosol-analysis-32-320.jpg)























































































![5G Explained! A High Level Overview [Introduction]](https://cdn.slidesharecdn.com/ss_thumbnails/5gexplainedahighleveloverview-260119165306-cc137a3e-thumbnail.jpg?width=640&height=640&fit=bounds)
















