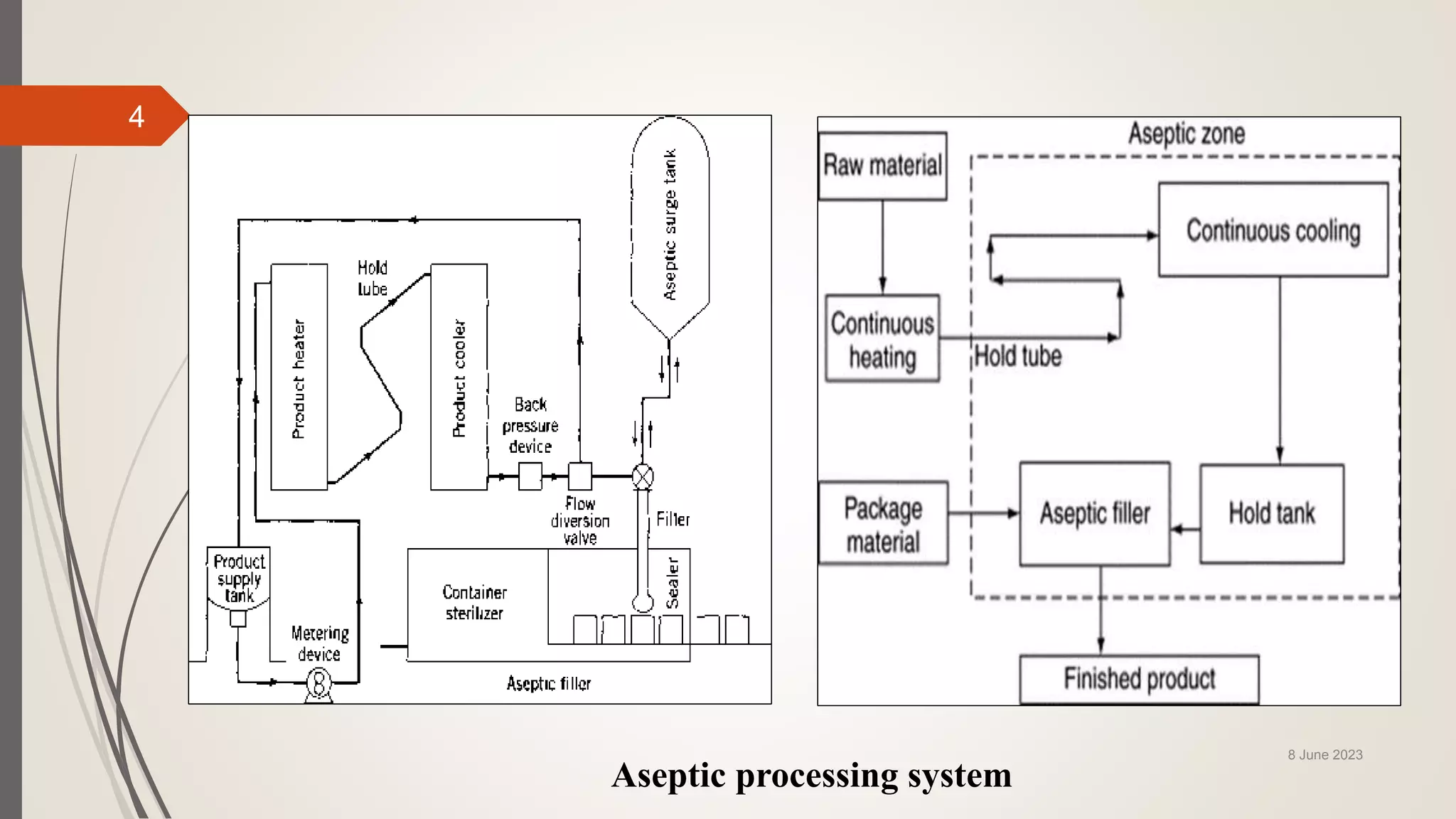
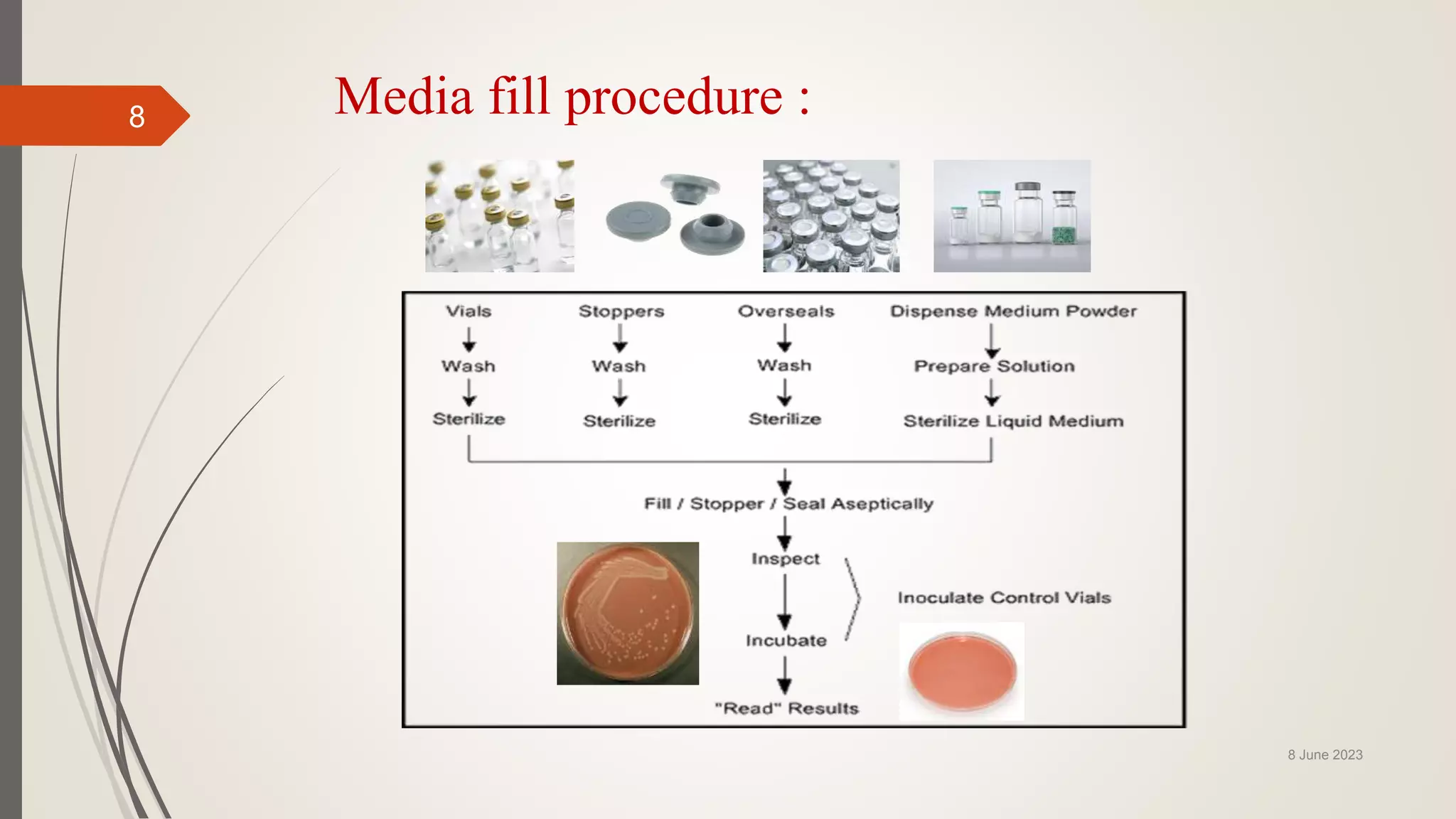
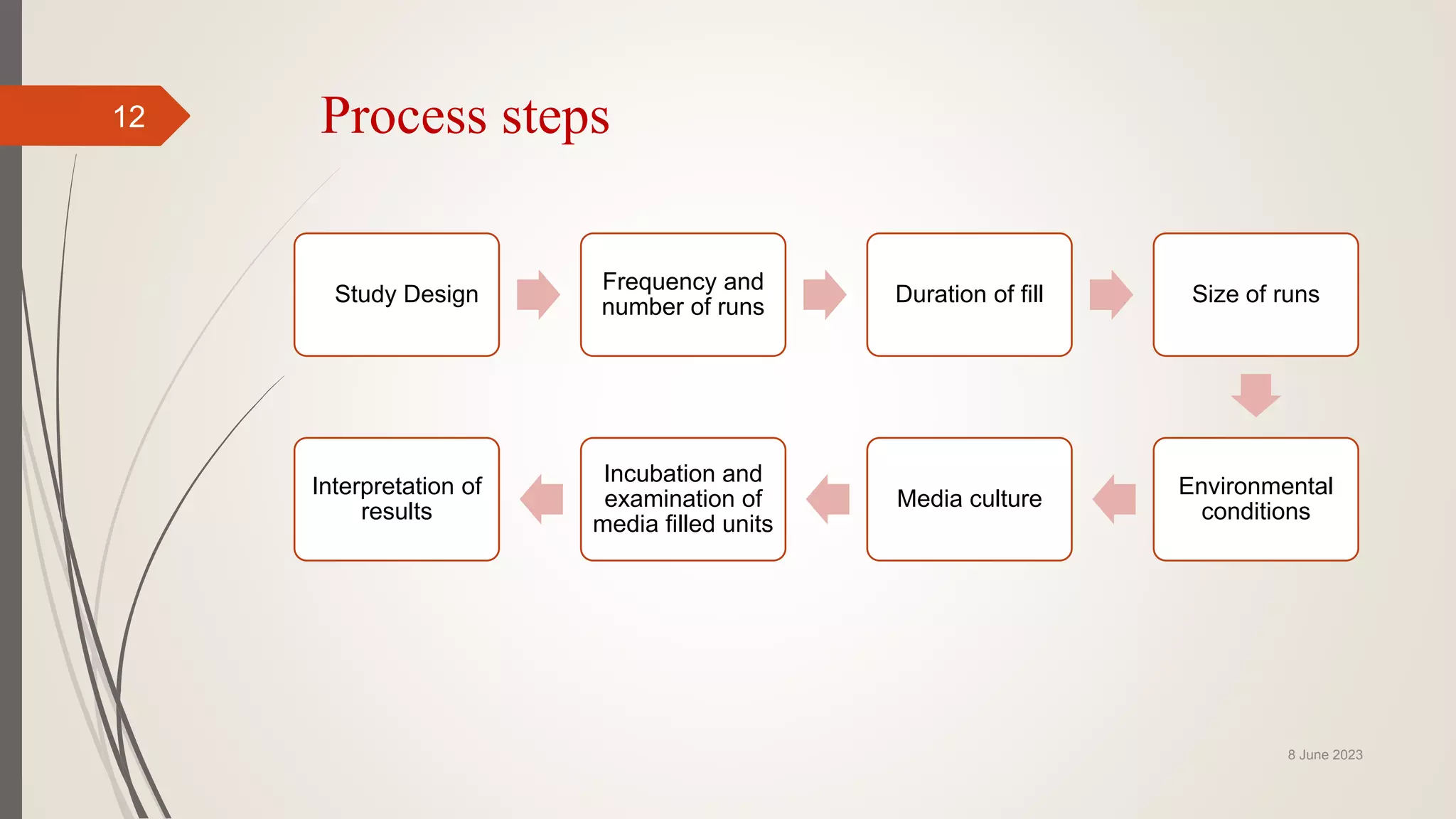
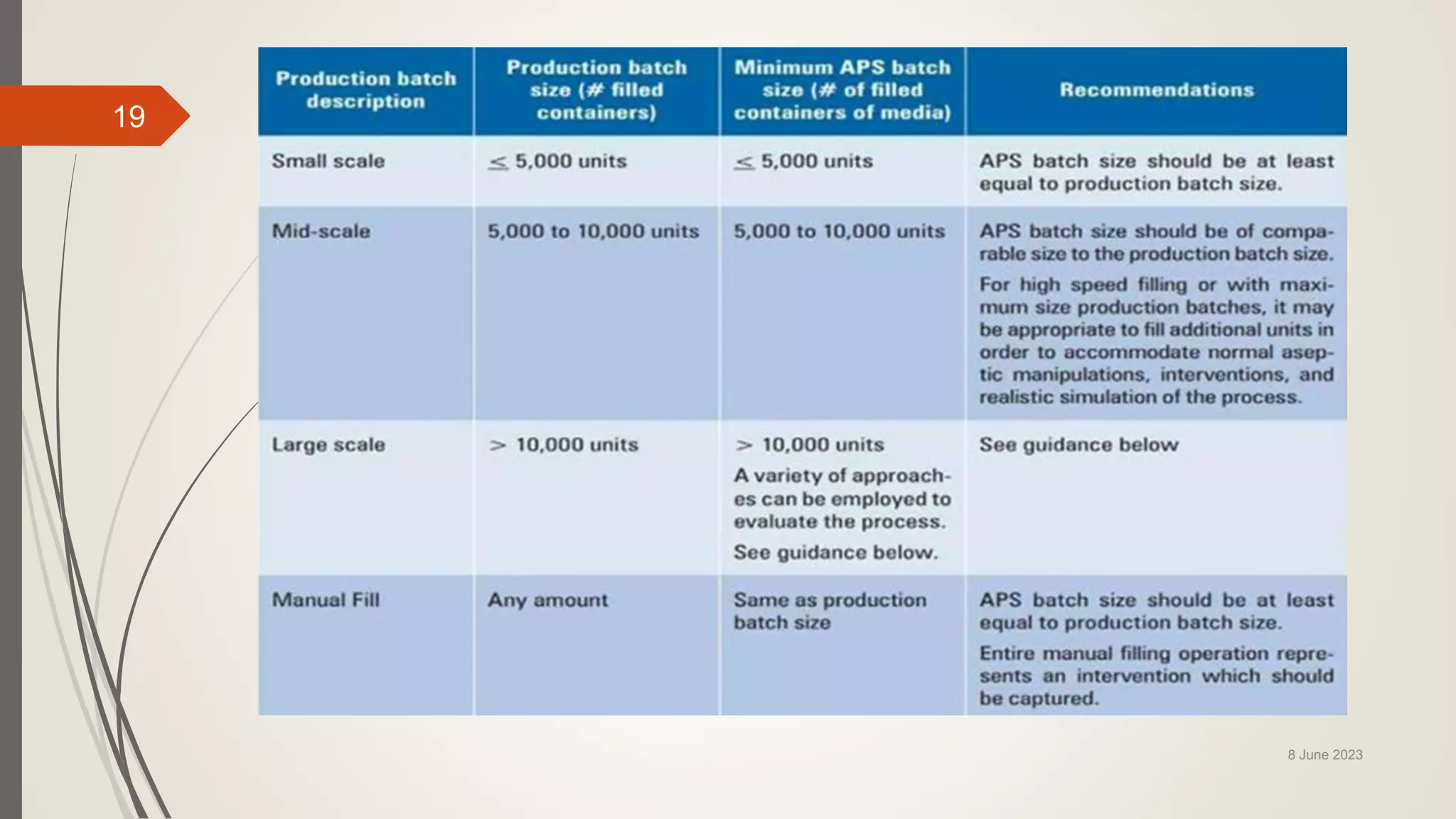
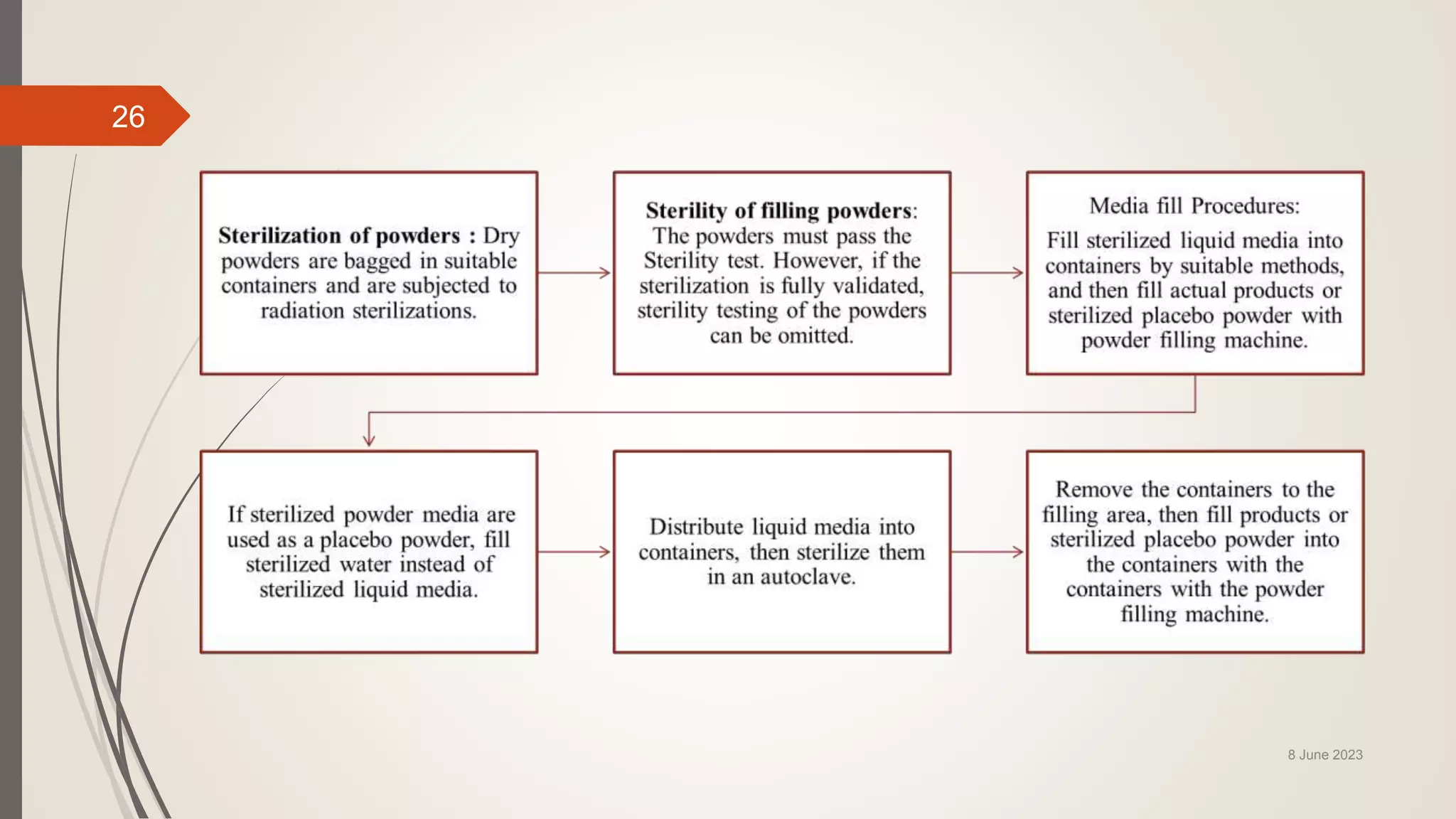
The document outlines aseptic media fill validation processes essential for ensuring the sterility of pharmaceutical products through controlled aseptic processing. It details the objectives, protocols, procedures, and standards for conducting media fills, including various validation techniques and environmental monitoring. Comprehensive guidelines on study designs, acceptance criteria, data documentation, and revalidation procedures are also presented to maintain compliance with good manufacturing practices.