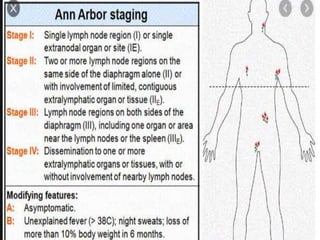
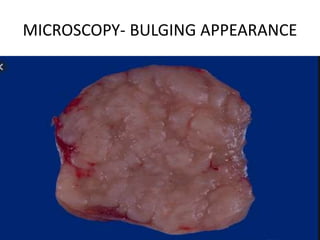
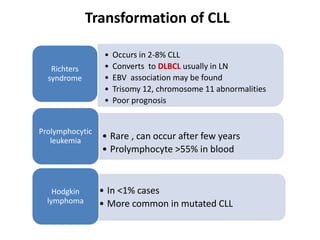
- A 55-year-old male presented with peripheral and central lymphadenopathy and splenomegaly but was asymptomatic. A lymph node biopsy was performed.
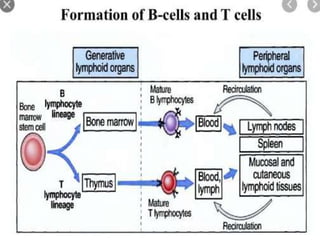
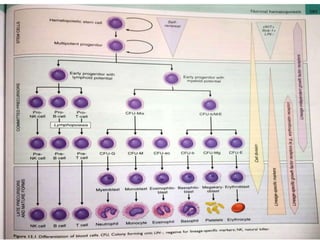
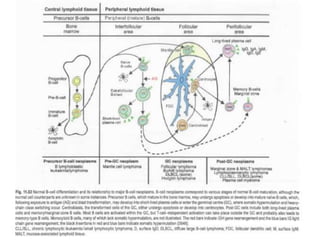
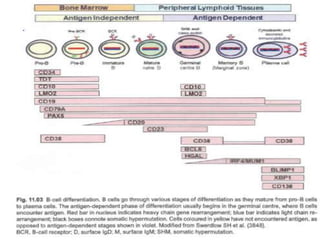
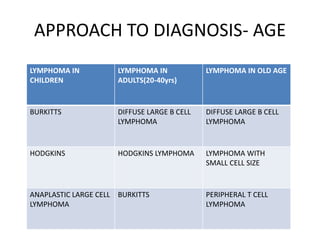
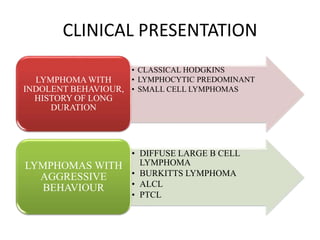
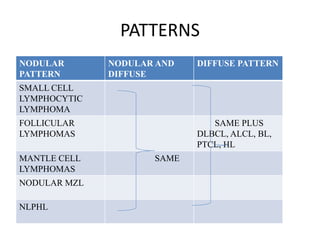
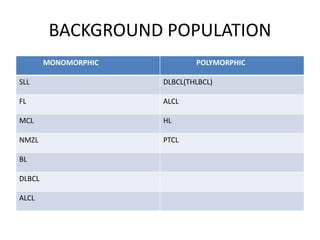
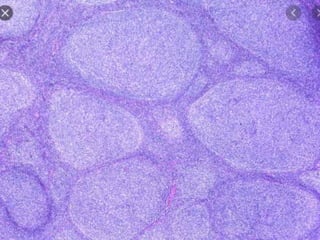
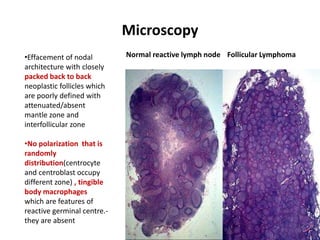
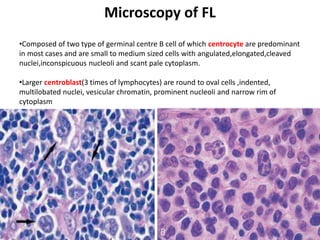
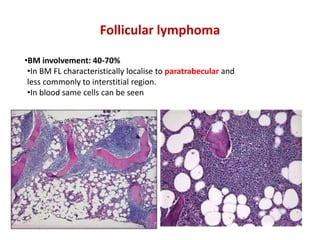
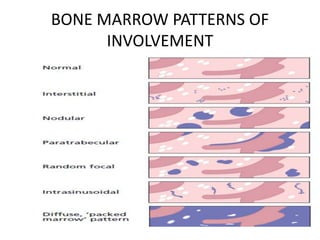
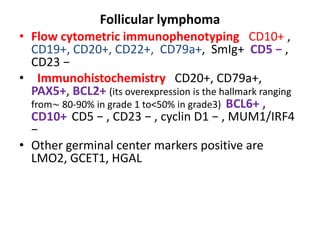
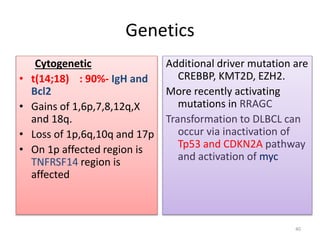
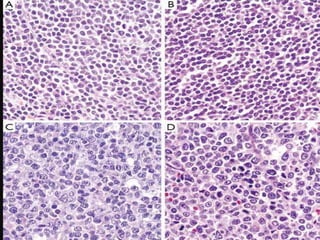
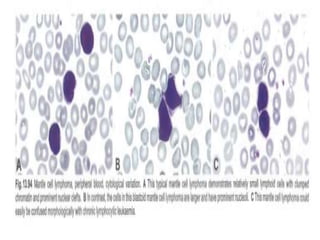
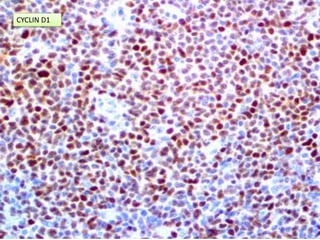
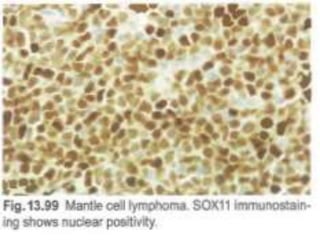
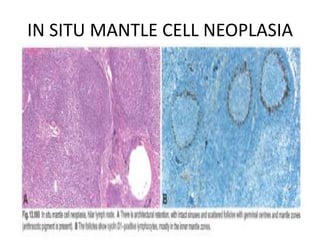
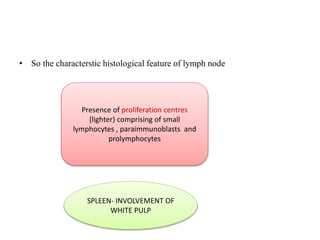
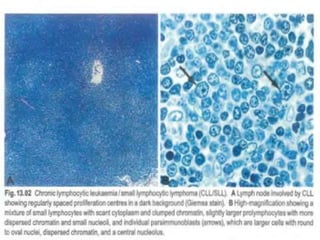
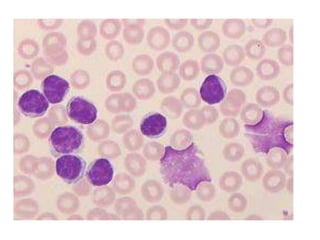
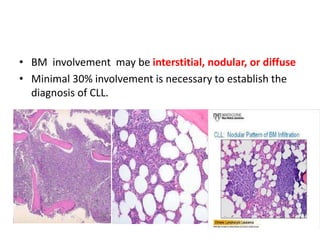
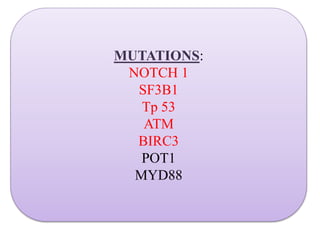
- Microscopic examination revealed features consistent with follicular lymphoma (FL), a neoplasm composed of follicle center B-cells which usually has at least a partially follicular pattern. FL is characterized by t(14;18) translocation and involves bone marrow in 40-70% of cases.
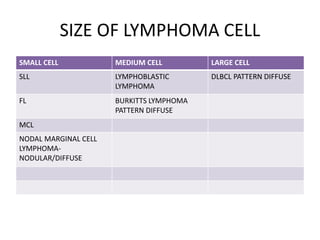
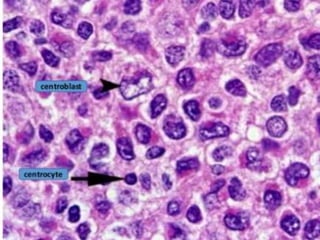
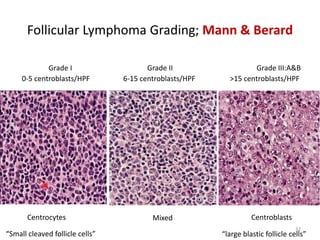
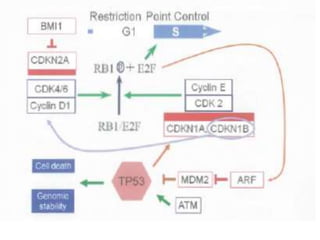
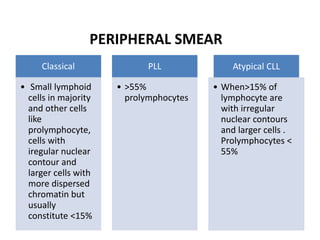
- FL was further classified based on the number of centroblasts per high power field according to the Mann and Berard grading system into Grade I (0-5 centroblasts/HPF), Grade II (6-15 centroblasts/

























































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)





