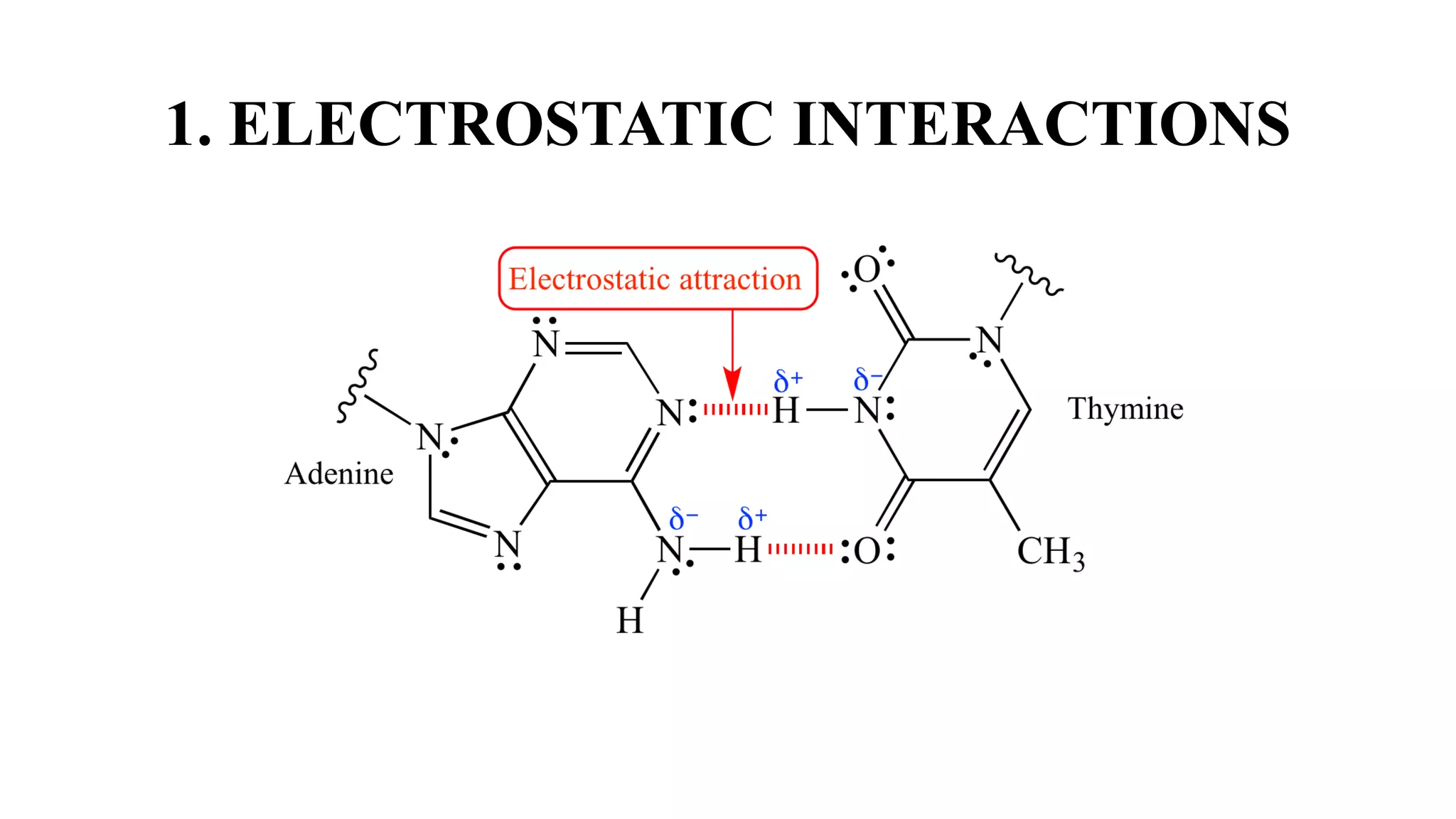
The document discusses electrostatic interactions and methods for predicting protein-nucleic acid interactions. It covers the role of electrostatics in determining biomolecular structure and interactions. It also describes Poisson-Boltzmann theory as a framework for modeling electrostatics and various software tools that solve the Poisson-Boltzmann equation. Finally, it outlines different approaches for modeling and predicting protein-nucleic acid interactions, including molecular dynamics simulations and statistical and knowledge-based potential functions.