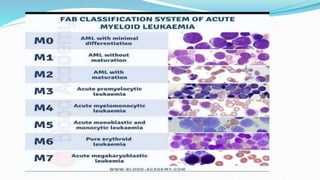
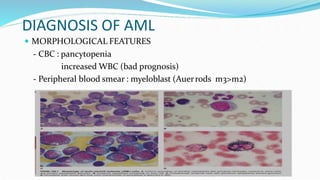
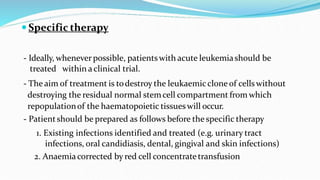
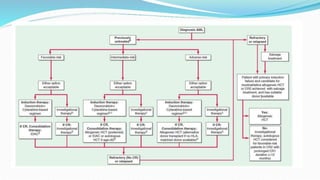
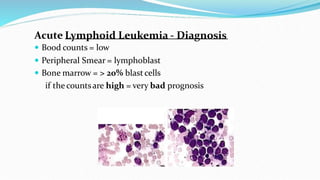
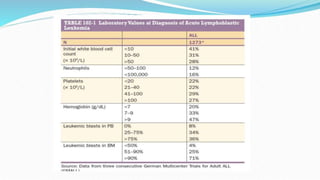
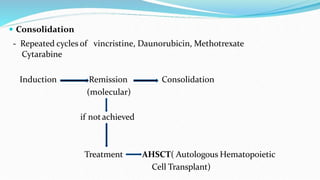
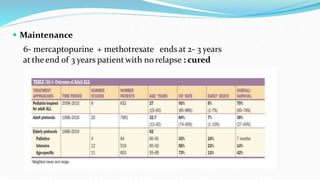
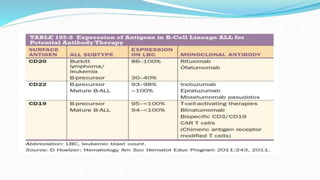
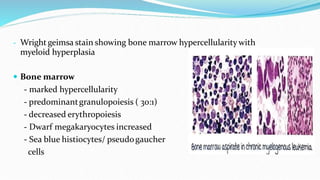
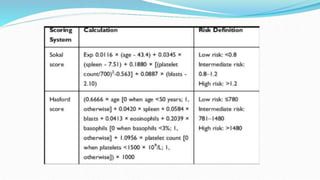
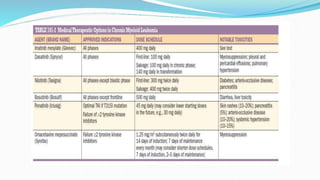
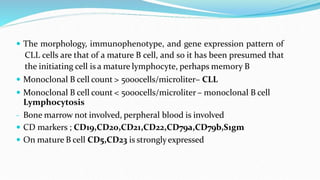
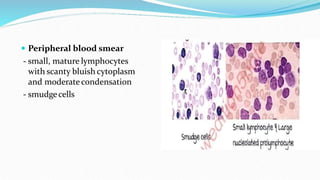
This document discusses diagnosis and management of various types of leukemia. It provides information on acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and chronic myeloid leukemia (CML). For AML, it describes diagnostic criteria including blood tests, bone marrow examination, immunophenotyping, cytogenetics and molecular genetics. It outlines treatment including induction chemotherapy and consolidation therapy. For ALL, it similarly discusses diagnosis and treatment approaches including chemotherapy, immunotherapy and stem cell transplantation. Finally, it covers CML including the Philadelphia chromosome abnormality and treatment with tyrosine kinase inhibitors like imatinib.








































































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)
















