Downloaded 178 times



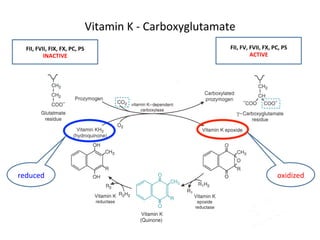
![Vitamin
K
Deficiency
• Found
in
leafy
green
plants
as
phylloquinone
and
in
bacteria
as
menaquinone
• Required
for
the
a]achment
of
gamma-‐carboxyglutamic
acid
(GLA)
residues
to
the
VKDFs
• Factors
produced
in
the
absence
of
VK
lack
the
required
number
of
GLA
residues
and
are
func<onally
inac<ve
à
PIVKAs
• GLA
residues
facilitate
the
a]achment
of
the
factors
to
PF3
through
calcium
binding
• VK
deficiency
seen
in
1. Absence
of
bile
salts
in
GI
tract
• VK
is
fat
soluble
à
bile
salts
are
required
for
adsorp<on
2. Malabsorp<on
syndromes
• VK
is
absorbed
primarily
through
the
GI
tract
3. Dietary
lack
of
phylloquinone
• Due
to
lack
of
green
leafy
vegetables
in
the
diet
4. An<bio<c
therapy
• Kills
the
normal
flora
of
the
GI
tract—responsible
for
menaquinone
5. Bowel
surgery
• Combina<on
of
loss
of
phylloquinone
and
menaquinone
6. Newborn
infants
• Deficient
in
vitamin
K
at
birth
3](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-3-320.jpg)






![Bleeding
disorders
have
been
recognized
since
ancient
<mes…
The
Talmud
(2nd
century
AD)
states
that
male
babies
do
not
have
to
be
circumcised
if
two
brothers
have
died
from
the
procedure
In
12th
century
Albucasis,
an
Arab
physician,
wrote
about
a
family
in
which
males
died
of
excessive
bleeding
from
minor
injuries
In
1803,
Dr.
John
O]o,
Philadelphia,
wrote
about
an
inherited
hemorrhagic
disposi<on
affec<ng
males
In
1828
at
the
University
of
Zurich,
“hemophilia"
was
first
used
to
describe
a
bleeding
disorder](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-13-320.jpg)


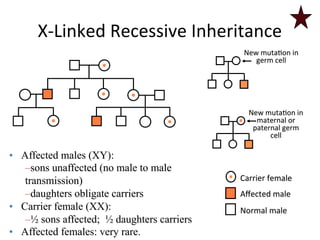
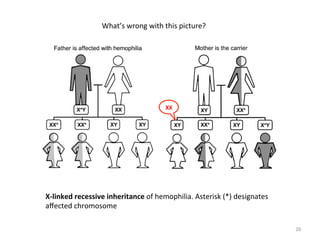
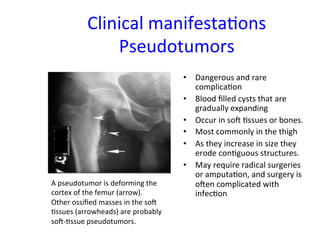
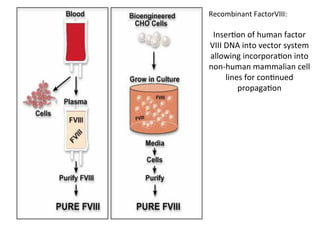
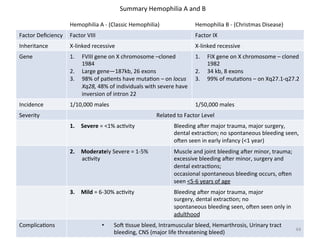
![Disorders
of
Secondary
Hemostasis
• Hemophilia
A
and
B
– Sex-‐linked
recessive
disorders
first
described
in
the
Talmud
in
the
5th
century
– By
the
end
of
the
19th
century
the
cloZng
<mes
of
plasma
from
persons
with
hemophilia
were
found
to
be
greatly
prolonged
compared
with
the
cloZng
<mes
in
nonbleeders
– By
1947
hemophilia
was
a]ributed
to
a
single
protein
deficiency
– Pavlovsky
showed
that
plasma
of
some
hemophilic
pa<ents
could
correct
the
in
vitro
or
in
vivo
defects
of
other
pa<ents
with
clinically
iden<cal
bleeding
disorders
à
led
to
recogni<on
of
mulLple
types
of
hemophilia
– Hemophilias
A
and
B
together
occur
in
about
1/5,000
of
the
general
popula<on
– Hemophilia
A
is
about
4-‐6x
more
common
than
Hemophilia
B
– Defect
in
hemophilia
is
due
to
a
muta<on
located
on
the
“X”
chromosome
• Females
can
be
carriers
– One
normal
+
one
defecLve
“X”
chromosome
• Females
are
asymptoma<c
1. Transmit
one
abnormal
X
chromosome
to
each
male
offspring
2. Male
offspring
would
have
hemophilia
16](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-16-320.jpg)
![Disorders
of
Secondary
Hemostasis
• Hemophilia
A
and
B
– Sex-‐linked
recessive
disorders
first
described
in
the
Talmud
in
the
5th
century
– By
the
end
of
the
19th
century
the
cloZng
<mes
of
plasma
from
persons
with
hemophilia
were
found
to
be
greatly
prolonged
compared
with
the
cloZng
<mes
in
nonbleeders
– By
1947
hemophilia
was
a]ributed
to
a
single
protein
deficiency
– Pavlovsky
showed
that
plasma
of
some
hemophilic
pa<ents
could
correct
the
in
vitro
or
in
vivo
defects
of
other
pa<ents
with
clinically
iden<cal
bleeding
disorders
à
led
to
recogni<on
of
mulLple
types
of
hemophilia
– Hemophilias
A
and
B
together
occur
in
about
1/5,000
of
the
general
popula<on
– Hemophilia
A
is
about
4-‐6x
more
common
than
Hemophilia
B
– Defect
in
hemophilia
is
due
to
a
muta<on
located
on
the
“X”
chromosome
• Females
can
be
carriers
– One
normal
+
one
defecLve
“X”
chromosome
• Females
are
asymptoma<c
1. Transmit
one
abnormal
X
chromosome
to
each
male
offspring
2. Male
offspring
would
have
hemophilia
17](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-17-320.jpg)

![Thrombin
Genera<on
in
Normal
Individuals
• Normal
individuals
1. Forma<on
of
TF/VIIa
complex
following
vascular
injury
2. Extrinsic
pathway
ac<va<on
of
FX
via
Extrinsic
Tenase
complex
[TF:FVIIa:PF3:Ca2+]
3. Ini<al
burst
of
thrombin
4. TFPI
is
released
from
endothelial
cells
and
down-‐regulates
the
[TF:VIIa:FXa]
complex
à
turns
off
the
extrinsic
genera<on
of
thrombin
5. Thrombin
generated
from
the
Extrinsic
Pathway
à
thrombin
genera<on
6. Thrombin
converts
FVIII
à
FVIIIa
7. FVIIIa
is
a
cofactor
for
the
forma<on
of
the
Intrinsic
Tenase
complex
[VIIIa:IXa:PF3:Ca2+]
8. Intrinsic
tenase
complex
is
responsible
for
con$nued
thrombin
genera<on](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-21-320.jpg)
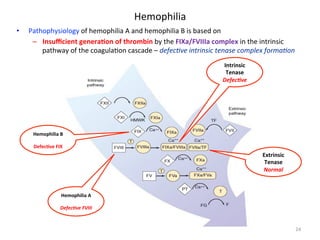
![Pathophysiology
Hemophilia
A
• Insufficient
genera<on
of
thrombin
by
– FIXa/VIIIa
complex
through
the
intrinsic
pathway
of
coagula<on
cascade
– Bleeding
severity
complicated
by
excessive
fibrinolysis
1. IIa
cannot
feedback
to
ac<vate
VIII
à
VIIIa—VIII
is
defec3ve
[Hemophilia
A]
2. As
a
result
—VIIIa
cannot
bind
to
FIX
(FIX
is
normal
but
nonfunc3onal)
3. Due
to
lack
of
thrombin
ac<va<on
of
TAFI
– IIa
à
genera<on
of
TAFI
– In
normal
TAFI
turns
OFF
fibrinolysis
– In
hemophilia
there
is
a
decrease
in
TAFI
so
TAFI
cannot
turn
off
fibrinolysis
» Decreased
cloZng
due
to
decreased
FVIII/FIX
» Increase
in
fibrinolysis](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-22-320.jpg)
![Pathophysiology
Hemophilia
B
• Insufficient
genera<on
of
thrombin
by
– FIXa/VIIIa
complex
through
the
intrinsic
pathway
of
coagula<on
cascade
– Bleeding
severity
complicated
by
excessive
fibrinolysis
1. IIa
feedbacks
to
ac<vate
VIII
à
VIIIa
—FVIIIa
serves
as
a
cofactor
to
orient
FIXa
in
forming
the
intrinsic
tenase
complex
2. As
a
result
—FIX
cannot
bind
form
the
intrinsic
tenase
complex
to
ac3vate
FX
[FIX
is
defec3ve]
– In
hemophilia
TAFI
cannot
turnoff
fibrinolysis
» Decreased
cloZng
due
to
decreased
FVIII/FIX
» Increase
in
fibrinolysis](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-23-320.jpg)
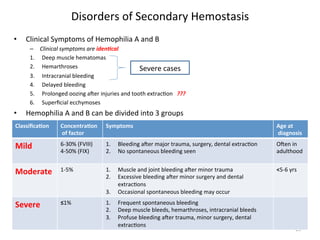

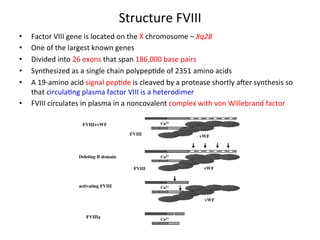
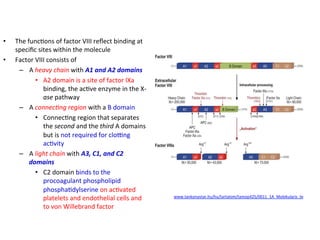
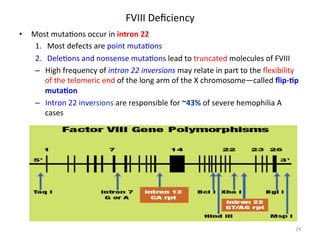
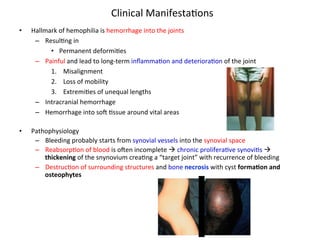
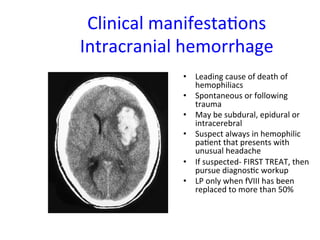











![FXIII
Deficiency
• FXIII
is
a
tetrameric
zymogen
that
is
converted
into
an
ac<ve
transglutaminase
by
thrombin
and
Ca2+
in
the
terminal
phase
of
the
cloZng
cascade
• Hallmarks
of
FXIII
deficiency
1. Umbilical
stump
bleeding
in
neonatal
period
2. Intracranial
hemorrhage
with
li]le
or
no
trauma
3. Recurrent
sow
<ssue
hemorrhage
4. Recurrent
spontaneous
abor<on
5. Impaired
wound
healing
and
spontaneous
abor<on
• Bleeding
– Usually
associated
with
trauma
– Bleeding
at
<me
of
surgery
is
not
excessive
• Delayed
bleeding
can
occur](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-49-320.jpg)

![Clinical Testing for Factor XIII
Urea
Clot
Solubility
Test
• Qualita<ve
assay
• Pa<ent
sample
is
clo]ed
and
then
clot
is
placed
in
5
M
urea
for
24
hours
at
room
temperature
– Clots
formed
by
normal
individuals
remain
stable
– Clots
from
factor
XIII
deficient
pa<ents
dissolve
• Detects
only
the
most
severely
affected
homozygous
pa<ents
with
1%
to
2%
factor
XIII
ac<vity
or
less
• Urea
solubility
assay
• Factor
XIII
forms
covalent
cross
links
between
fibrin
chains
• In
the
absence
of
Factor
XIII
à
the
fibrin
clot
will
be
dissolved
by
5
M
urea
which
disrupts
the
hydrogen
bonds
• This
assay
will
be
abnormal
only
if
the
factor
XIII
level
is
<2-‐5%](https://image.slidesharecdn.com/lecture6coagulationfall2014-141207132412-conversion-gate01/85/Lecture-6-coagulation-fall-2014-51-320.jpg)


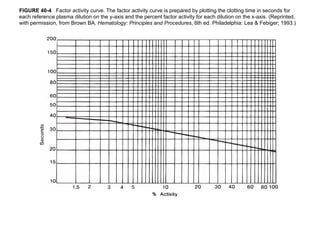
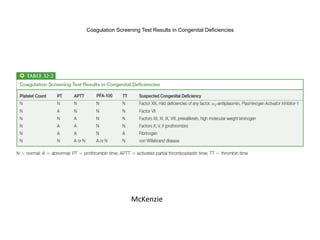

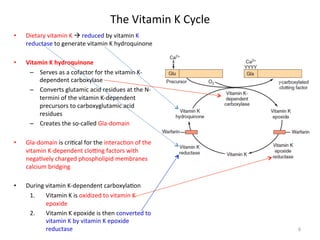
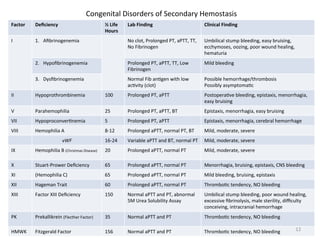
The document discusses congenital and acquired disorders related to secondary hemostasis, focusing on various coagulation factor deficiencies and their implications. Key points include the role of vitamin K in synthesizing coagulation factors, the impact of renal dysfunction on platelet function, and the specifics of hemophilia A and B. It also highlights the historical context and diagnostics of bleeding disorders and discusses treatment options for vitamin K deficiency and hemophilia.



































































































