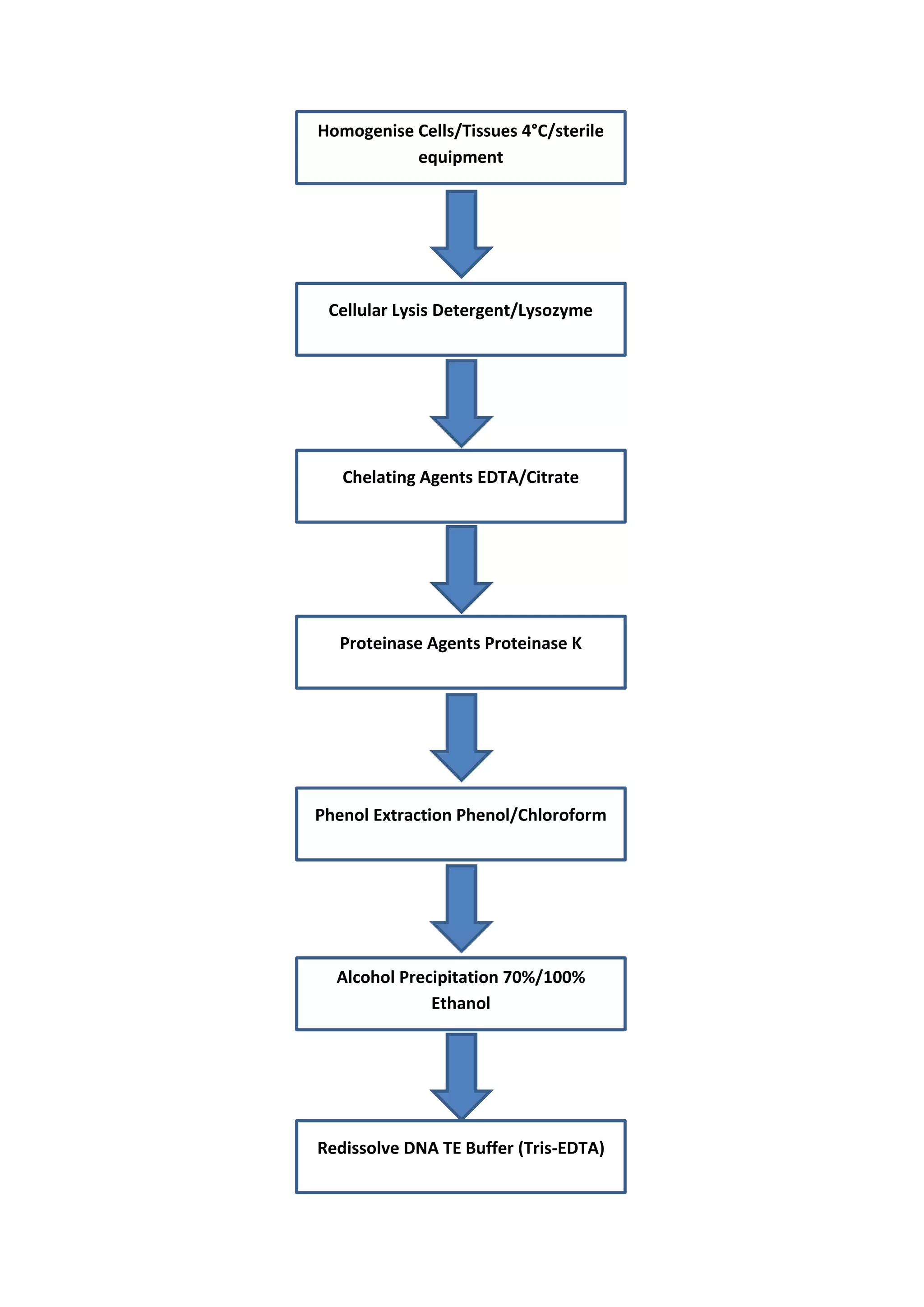
This document outlines methods for isolating and purifying DNA and RNA for analysis, emphasizing the importance of minimal mechanical disruption and the use of enzymatic treatments to prevent degradation. It details the steps involved, including cell lysis, contaminant removal, and precipitation of nucleic acids, as well as methods for assessing nucleic acid integrity and concentration. Special attention is given to the extraction of specific RNA types, such as eukaryotic mRNA, using affinity chromatography.