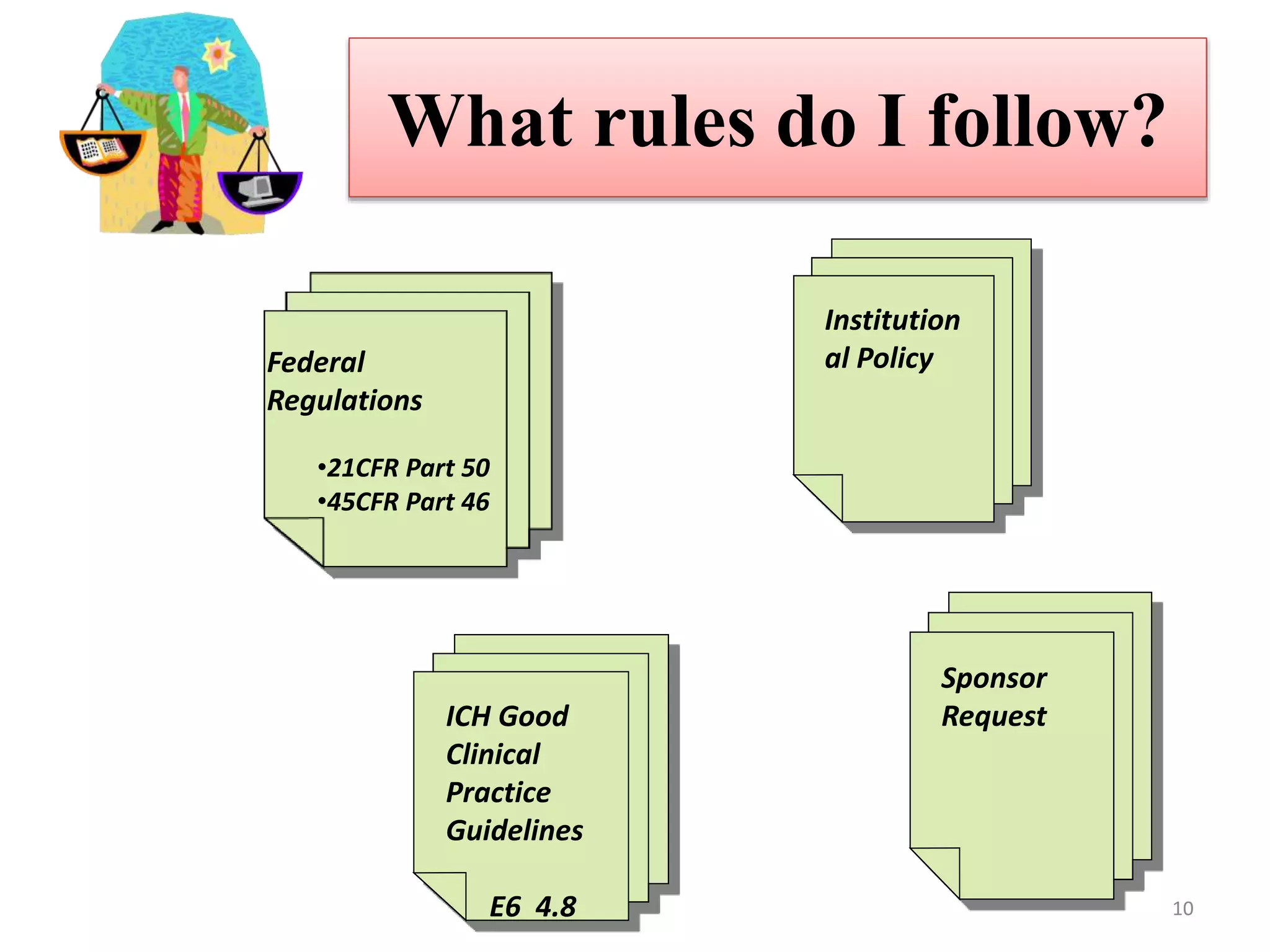
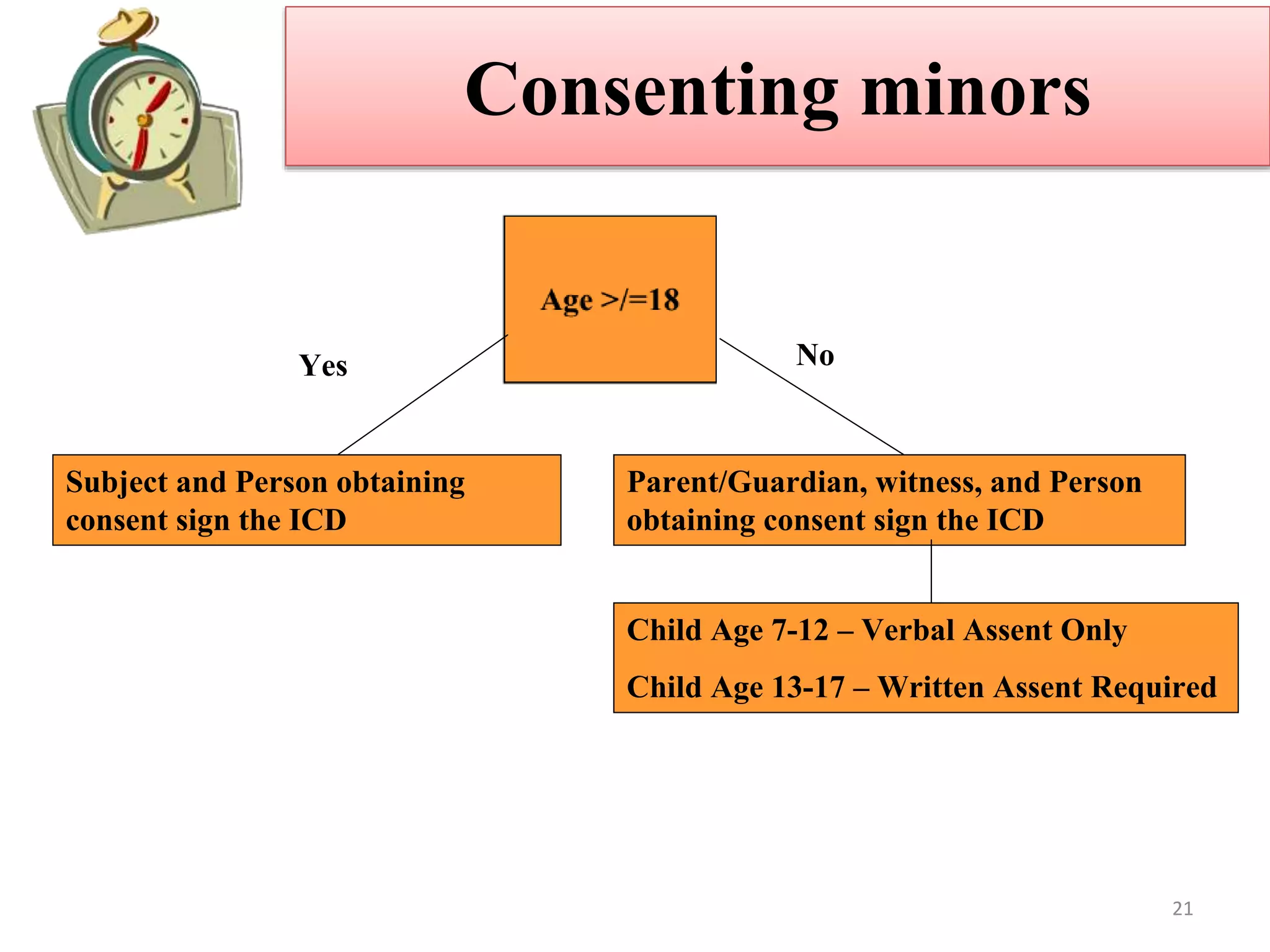
The document discusses informed consent, including its purpose, history, principles, elements, documentation, and common audit findings. Informed consent is a process of communicating with research participants to ensure they understand the study and can choose to participate voluntarily. It involves providing information on the study's risks and benefits and obtaining signed documentation of consent. Revisions are needed if the study procedures change, and special considerations apply for populations like children, non-English speakers, and the illiterate.