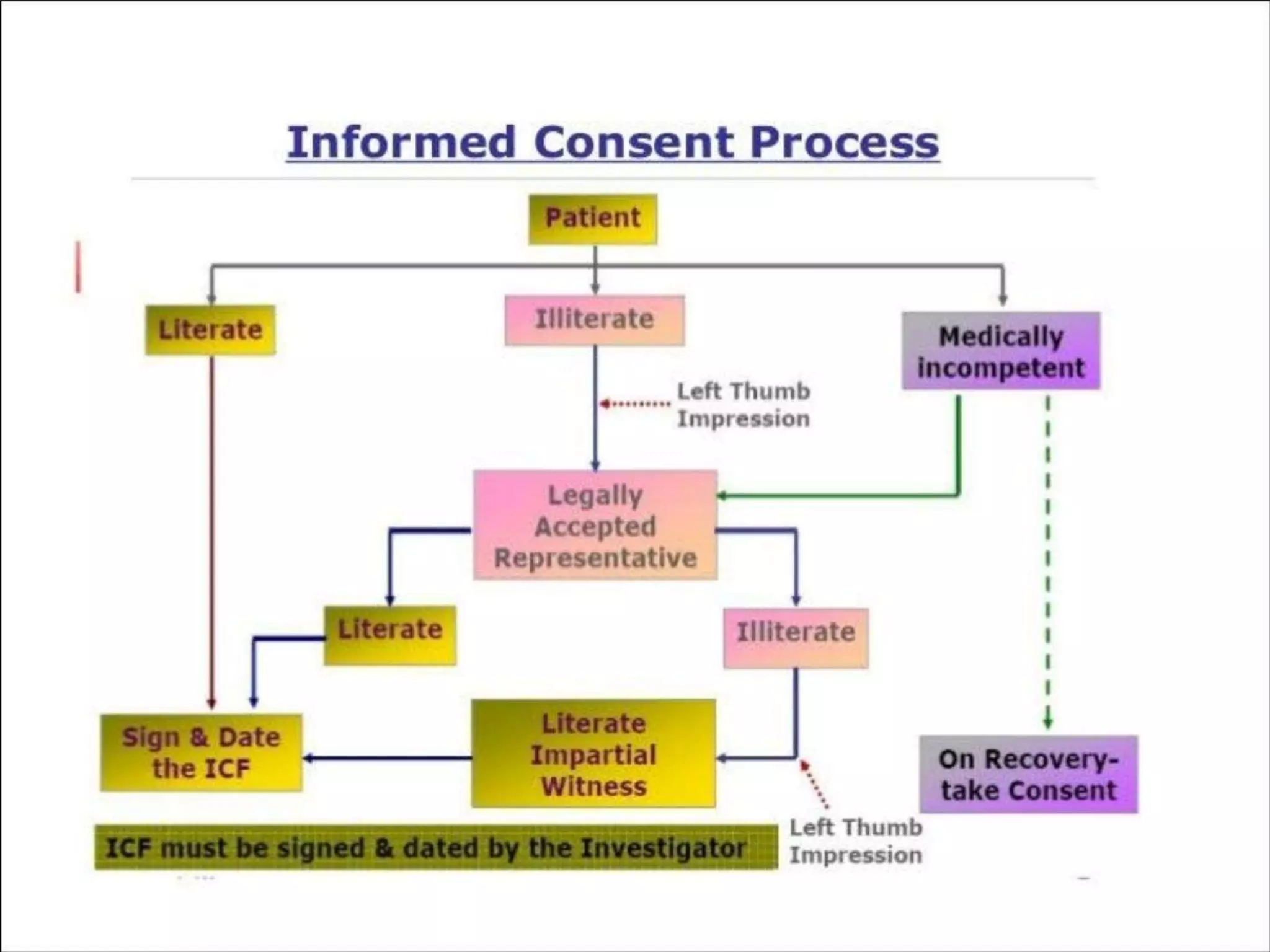
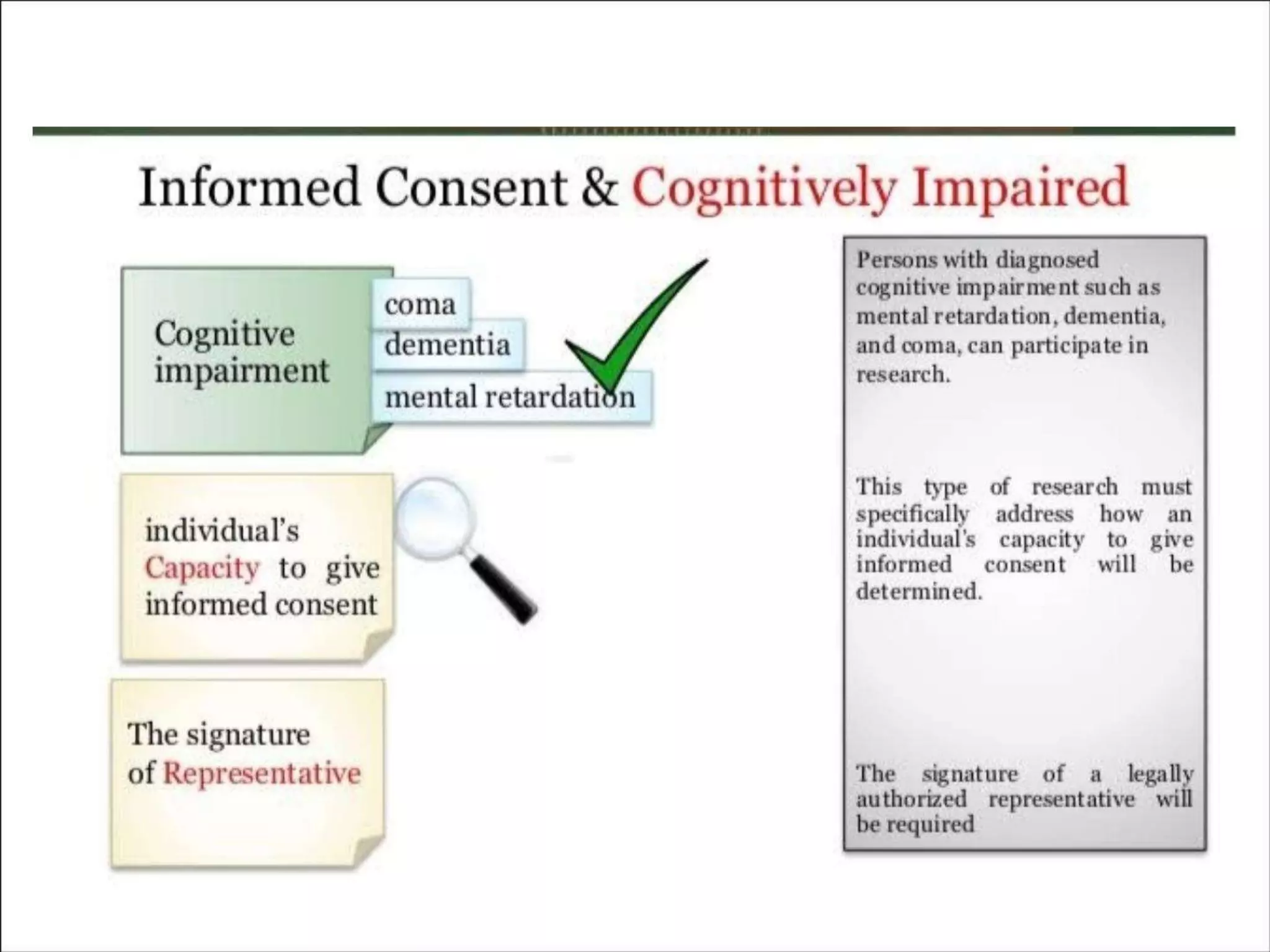
The document outlines the concept of informed consent, describing it as a communication process between researchers and participants, essential for ethical research practices. It includes historical examples of unethical research, basic principles of informed consent such as autonomy and beneficence, and specific considerations for different groups, including vulnerable populations. Additionally, it details the key elements required in informed consent forms and the necessity for revisions as new safety information arises.





![Nazi Prisoner Research During World War II:
Research Done The Wrong Way- Nazi Prisoner Research World War II
[1939-1945].
•World war II and unethical clinical trials done by US health services gave birth of bio-
ethics and creation of IRB and notion of informed consent](https://image.slidesharecdn.com/icfppt-230524103732-e53c00d2/75/Informed-consent-form-6-2048.jpg)

![AUTONOMY
•Autonomy means that each person should be given the respect, time and opportunity
necessary to make his or her own decision.
BENEFICENCE
Beneficence obligates the investigator to secure the well being of all study subjects. It
is the responsibility of the investigator to protect subject from harm, as well as ensure
that they experience the possible benefits of investigator.
[ key: Maximize possible benefits and minimize possible harm.]](https://image.slidesharecdn.com/icfppt-230524103732-e53c00d2/75/Informed-consent-form-8-2048.jpg)