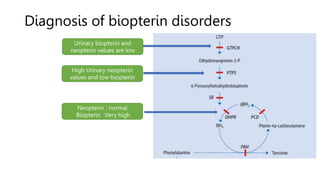
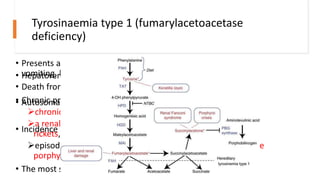
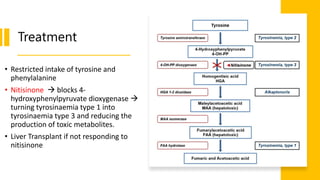
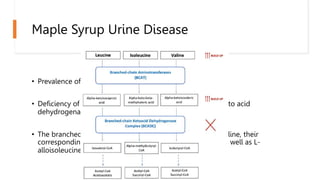
This document summarizes various inborn errors of amino acid metabolism, including:
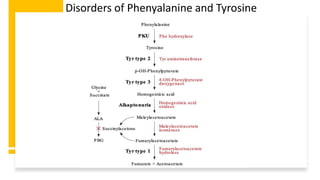
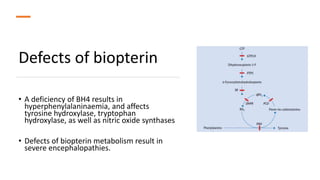
- Phenylketonuria (PKU), which results from a defect in the enzyme phenylalanine hydroxylase and can cause intellectual disability if left untreated;
- Tyrosinemias, including types 1, 2, and 3, which are caused by defects in tyrosine catabolism and can lead to liver or neurological complications;
- Alkaptonuria, which results from homogentisic acid dioxygenase deficiency and causes dark urine; and
- Maple syrup urine disease, caused by a branched-chain keto acid dehydrogenase complex defect leading to accumulation of leucine, isoleucine