Downloaded 381 times
![MEATABOLIC
INBORN
ERRORS
PRESENTER : #2013-02-095 / GIA K. SHARMA
INSTRUCTOR: DR. TOLUNIMI ADEDEJI [M.D.]
CENTRAL AMERICA HEALTH SCIENCE UNIVERSITY, BELIZE
July 10
2013
BIOCHEMISTRY
ASSIGNMENT](https://image.slidesharecdn.com/meatabolicinbornerrors-131117132757-phpapp01/85/Metabolic-inborn-errors-1-320.jpg)


![Defects of Lactate and others
- 20
Every child with unexplained
Neurological deterioration
Metabolic acidosis
Hypoglycemia
Inappropriate ketosis
Hypotonia
Cardiomyopathy
Hepatocellular dysfunction
Failure to thrive
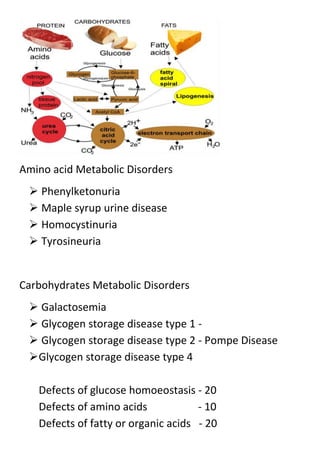
Phenylketonuria
Also known as PKU
Deficiency of Phenylalanine hydroxylase [PAH]](https://image.slidesharecdn.com/meatabolicinbornerrors-131117132757-phpapp01/85/Metabolic-inborn-errors-5-320.jpg)




![Diagnosis/Detection
To determine if someone has MSUD, you have to look the urine
odor for a sweet smell.
Blood test for amino acids.
If alloisoleucine is detected, the diagnosis is confirmed.
Deficiency of Branched chain alpha keto acid dehydrogenase
complex (BCKDC)
Sign and Symptoms- Maple syrup odor
Dehydration
Hypoglycemia
Ketoacidosis
Coma
Brain damage (if untreated)
Vomiting
Lethargy [lack of energy]
There is no cure for MSUD, however a special diet cant help
prevent these health problems.
Treatment- High doses of Thiamine](https://image.slidesharecdn.com/meatabolicinbornerrors-131117132757-phpapp01/85/Metabolic-inborn-errors-10-320.jpg)




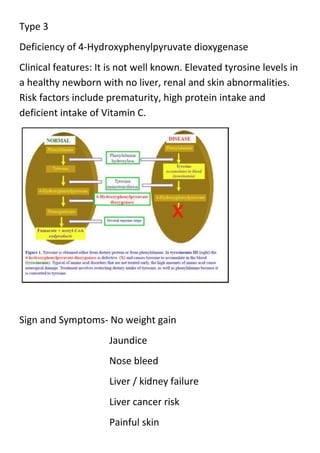
![Tyrosinemia
Mutations in the gene for fumarylacetoacetase
[FAH] result in enzyme that is not working well
or it is deficient.
It is a genetic disorder characterized by elevated blood
levels of the amino acids tyrosine.
It is also known as Hypertyrosinemia, type 1 and type 2 .
Type 1
Deficiency of Fumarylacetoacetate hydroxylase
Type 2
Deficiency of Tyrosine aminotransferase
Clinical features: Involve only skin, eyes and CNS which may
lead to excessive tearing, photophobia, eye pain and redness
and skin lesions.](https://image.slidesharecdn.com/meatabolicinbornerrors-131117132757-phpapp01/85/Metabolic-inborn-errors-15-320.jpg)







![Graves’ disease
It is a type of hyperthyroidism, which is caused by a generalized
over activity of the entire thyroid gland.
It is an autoimmune disease.
Cause : In this disease instead of destroying the thyroid gland,
an antibody called thyrotropin receptor antibody [TRAb] makes
the thyroid gland produce large amounts of thyroid harmone. It
is most common in women over age of 20 years.
Symptoms: Anxiety
Breast enlargement in men
Difficulty in concentrating
Double vision
Eyeballs that stick out [exophthalmos]
Eye irritation and tearing
Fatigue
Frequent bowel movements
Goiter [possible]
Increased appetite
Increased sweating
Insomnia
Muscle weakness
Nervousness
Weight loss
Restlessness](https://image.slidesharecdn.com/meatabolicinbornerrors-131117132757-phpapp01/85/Metabolic-inborn-errors-25-320.jpg)











This document provides information on various metabolic inborn errors including phenylketonuria, maple syrup urine disease, homocystinuria, tyrosinemia, galactosemia, glycogen storage diseases, and Niemann-Pick disease. It defines metabolic inborn errors as disorders caused by single gene defects that block metabolism. For each condition, it describes the genetic cause, signs and symptoms, diagnosis, and treatment. The document is presented as part of a biochemistry assignment on metabolic inborn errors for a health sciences university in Central America.




























































![Hypothalamus short ppt by Dr. Neha [PT].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/hypothalamusbydr-260124145759-b9f94a93-thumbnail.jpg?width=640&height=640&fit=bounds)


















