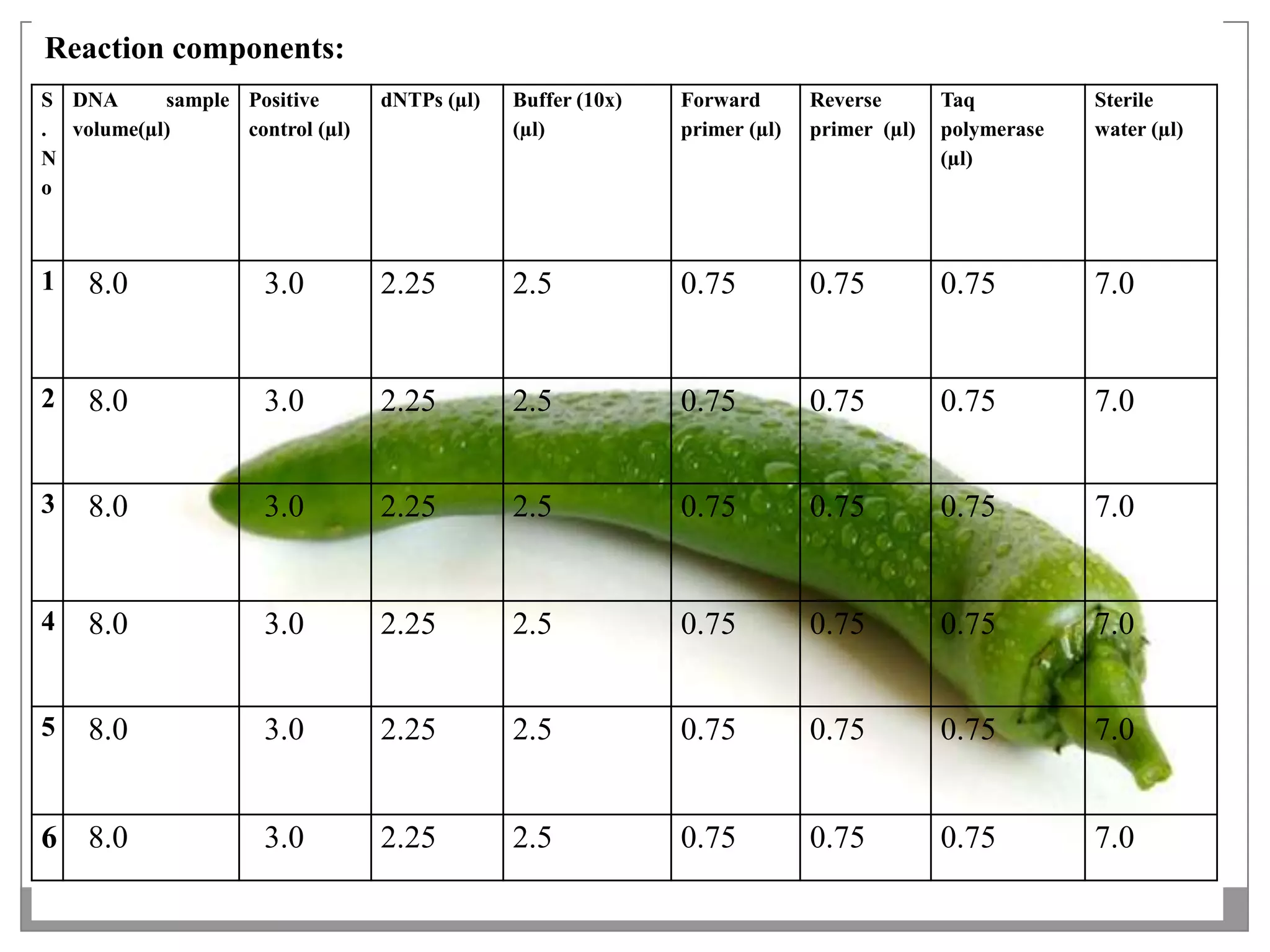
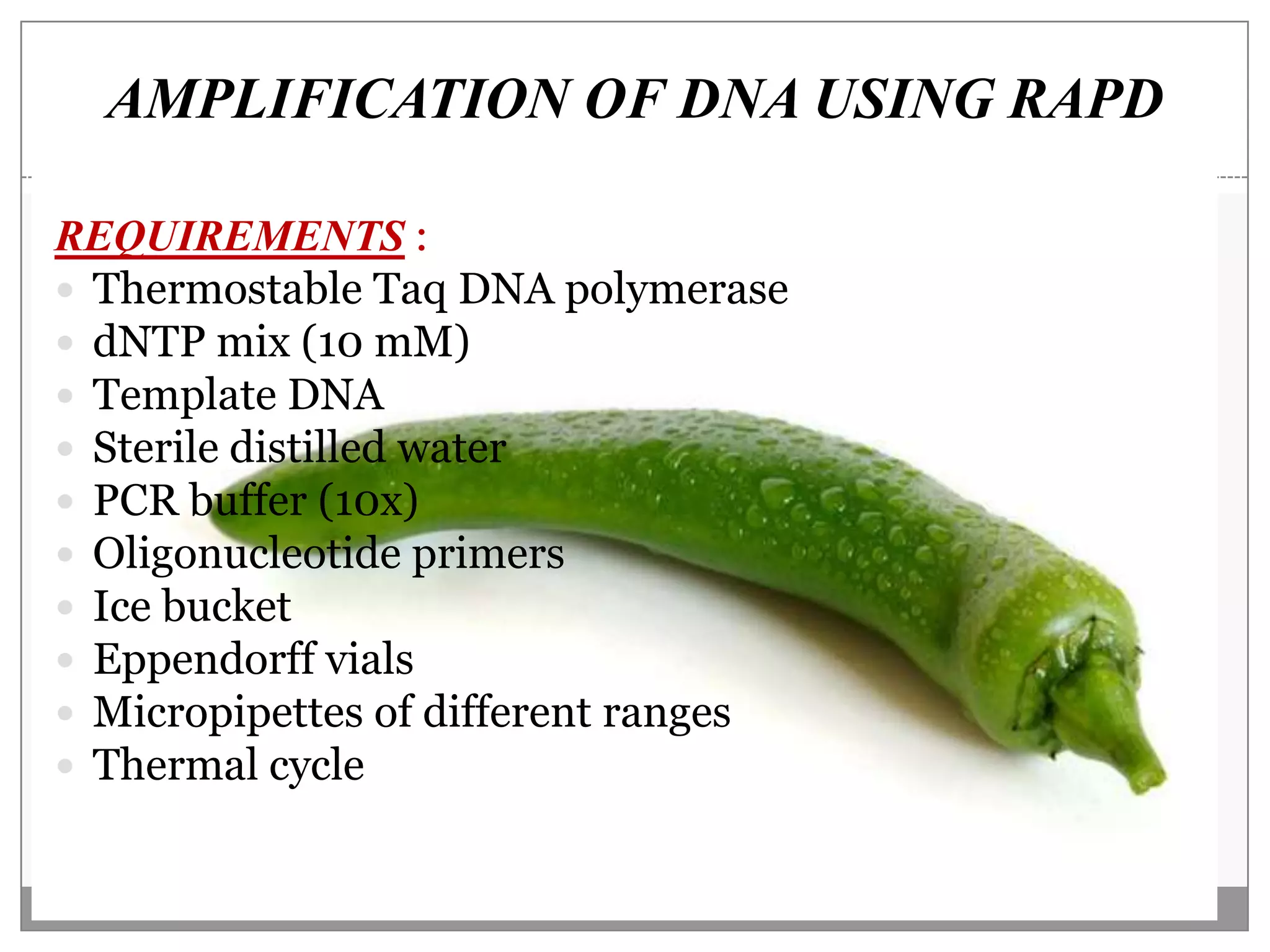
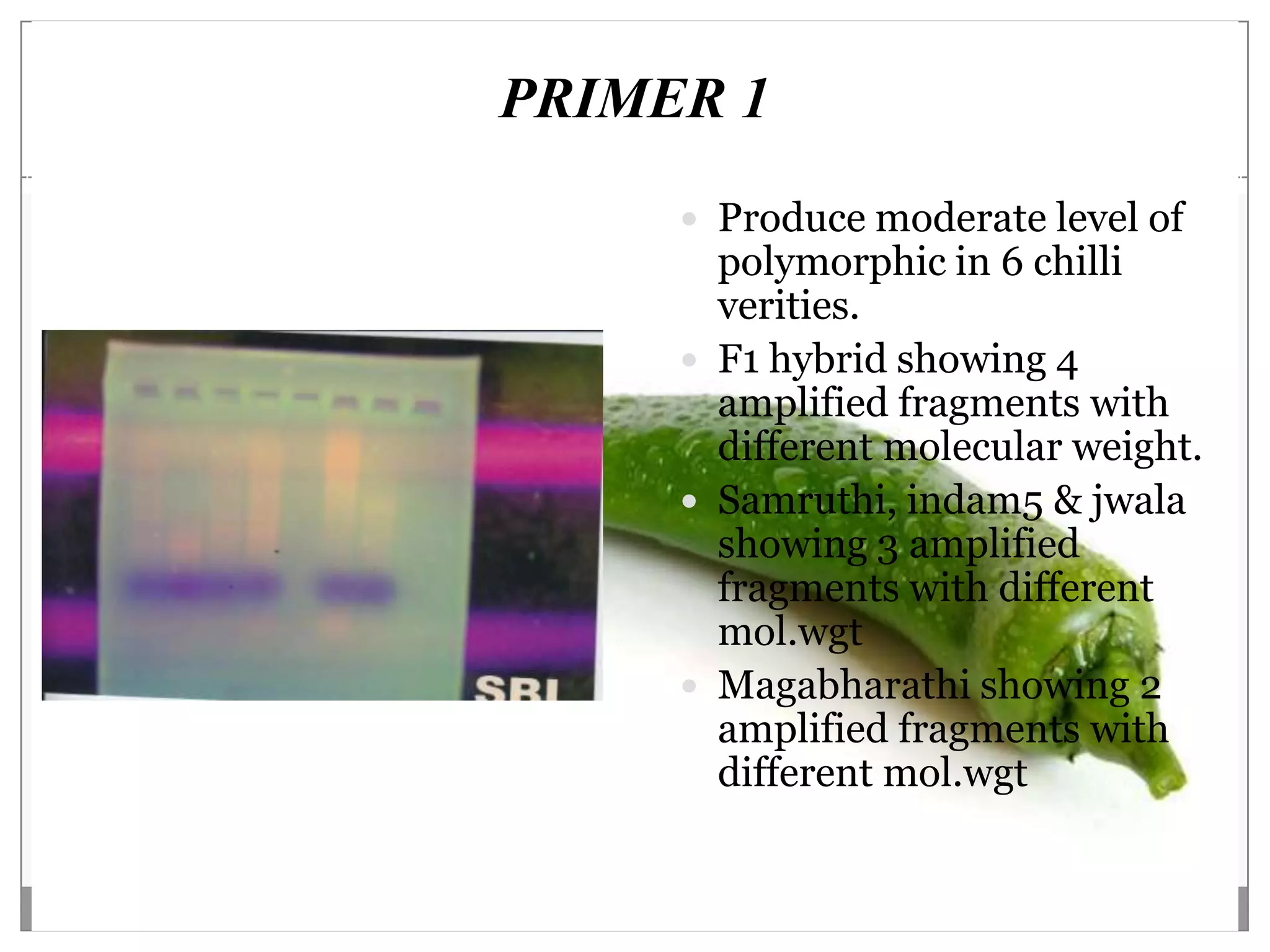
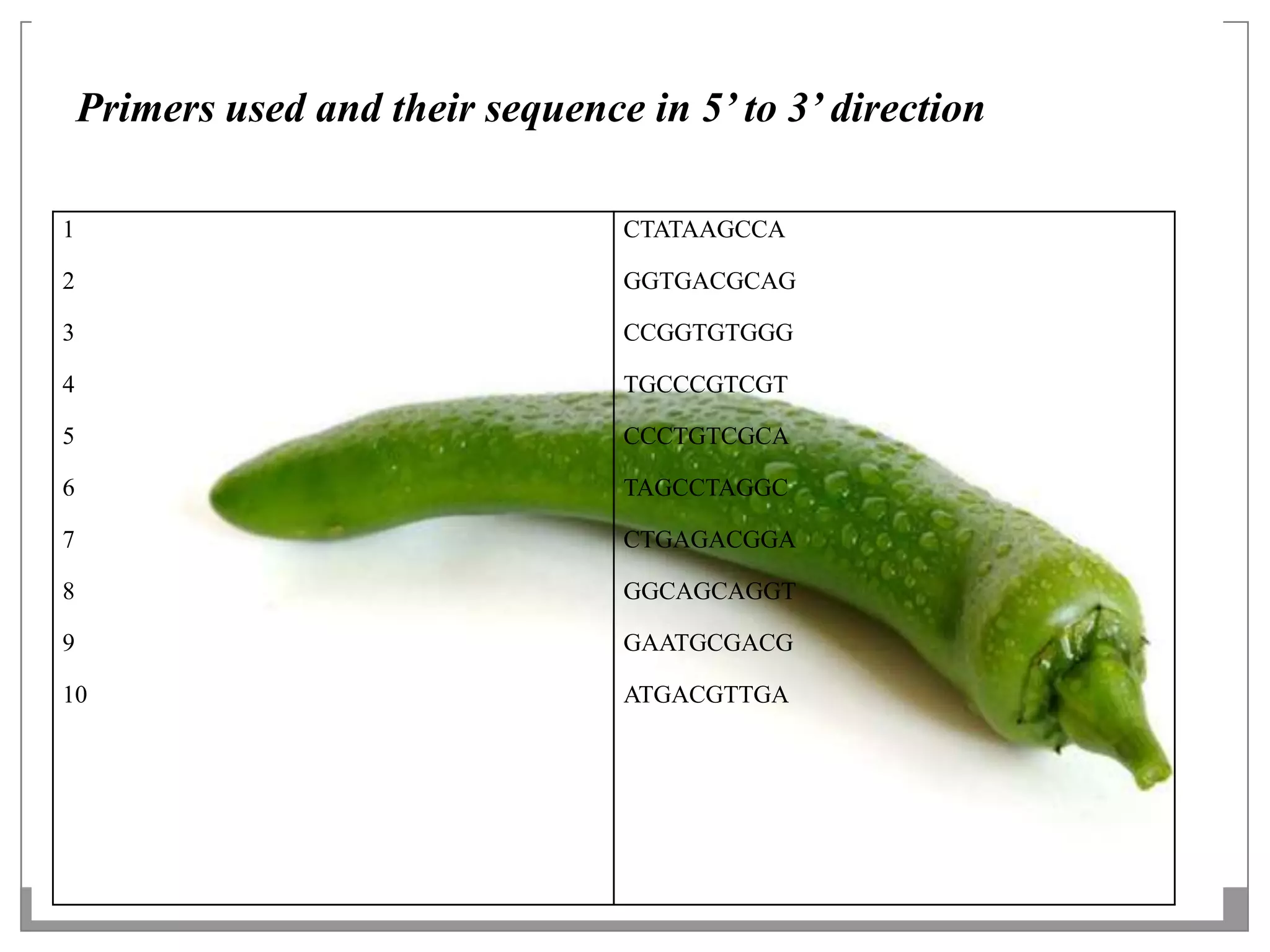
This document describes a study that aimed to identify polymorphism in different chilli varieties using DNA fingerprinting with RAPD. Seven chilli varieties were collected and genomic DNA was isolated from them using a CTAB method. Quality PCR was performed and DNA was amplified using RAPD. The PCR products were separated using urea polyacrylamide gel electrophoresis and visualized using silver staining. The study examined techniques for detecting genetic variation in chilli varieties at the DNA level.