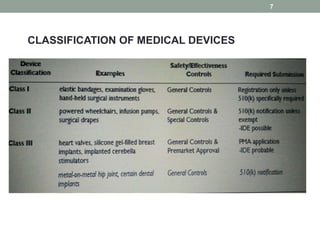
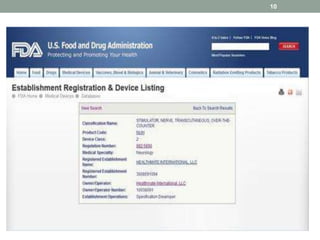
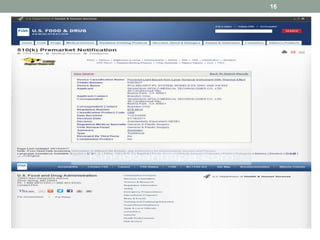
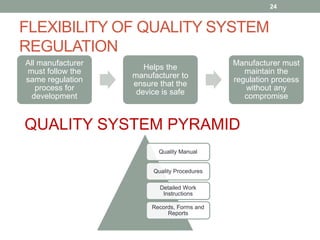
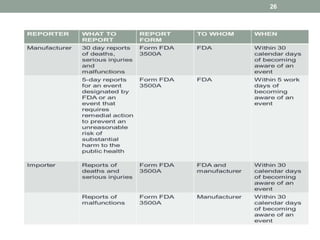
This document provides an overview of FDA regulation of medical devices in the United States. It defines key terms, describes the classification system for devices and corresponding levels of regulatory control. It outlines major premarket and postmarket requirements including establishment registration, 510(k) premarket notification, premarket approval, labeling, quality system regulation, medical device reporting and complaint handling. Major sections cover classification, premarket submissions, labeling and other compliance requirements enforced by the FDA to ensure device safety and effectiveness.


















































































![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)


![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)






