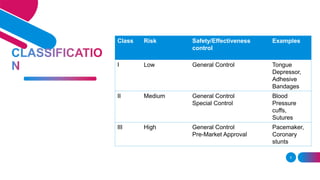
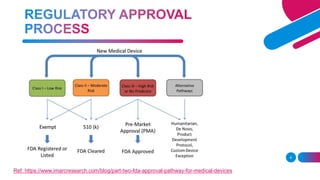
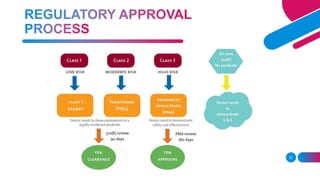
The document discusses the regulatory framework for medical devices in the USA, primarily focusing on the FDA's classification, approval processes, and the requirements for pre-market notifications and approvals for different classes of medical devices. It also details the investigational device exemption (IDE), quality system requirements, labeling mandates, post-market surveillance, and the unique device identification system. The document emphasizes the importance of ensuring safety and effectiveness through stringent regulatory compliance while providing an overview of in vitro diagnostics and their respective classifications.








































![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)
























































