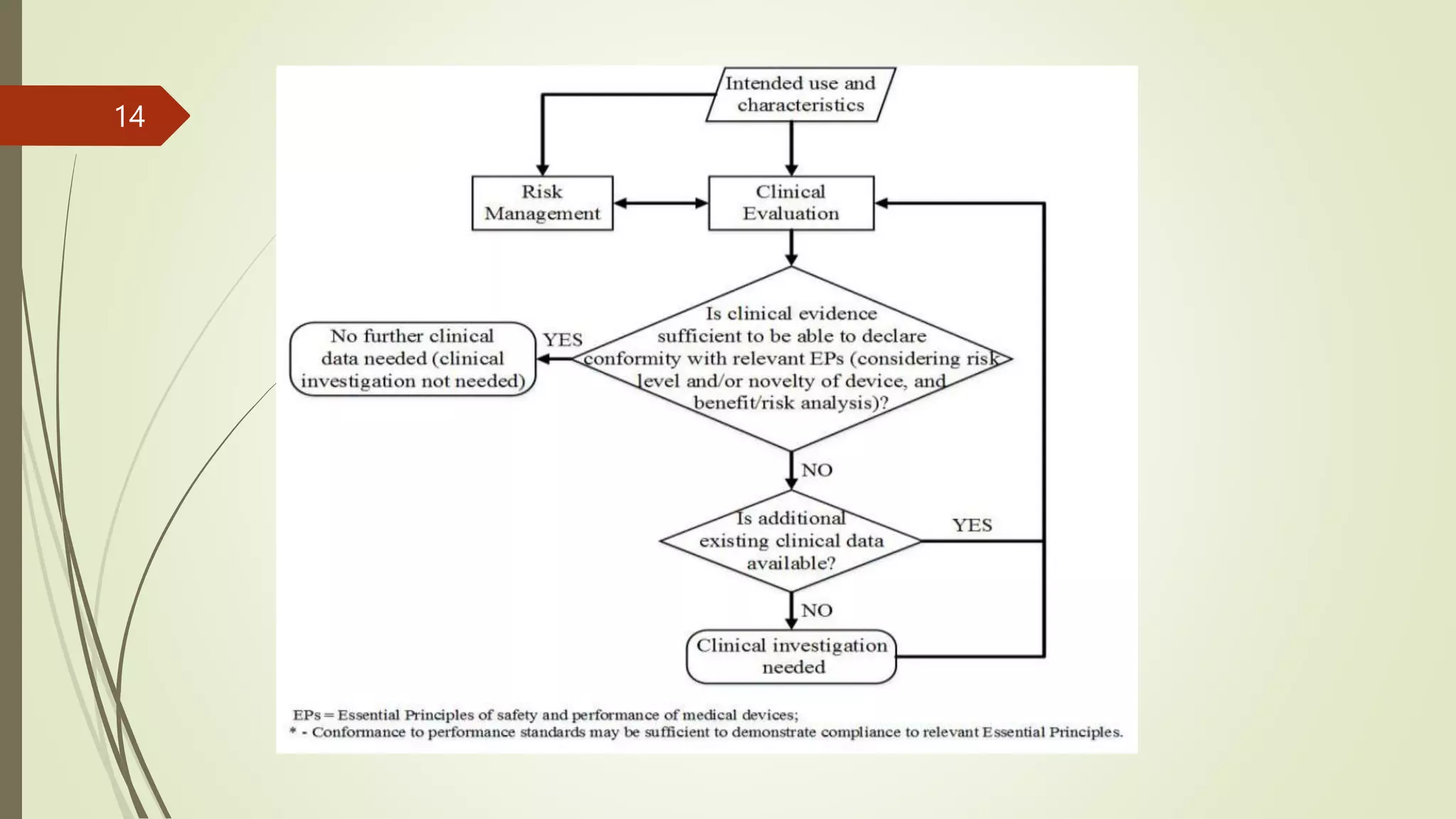
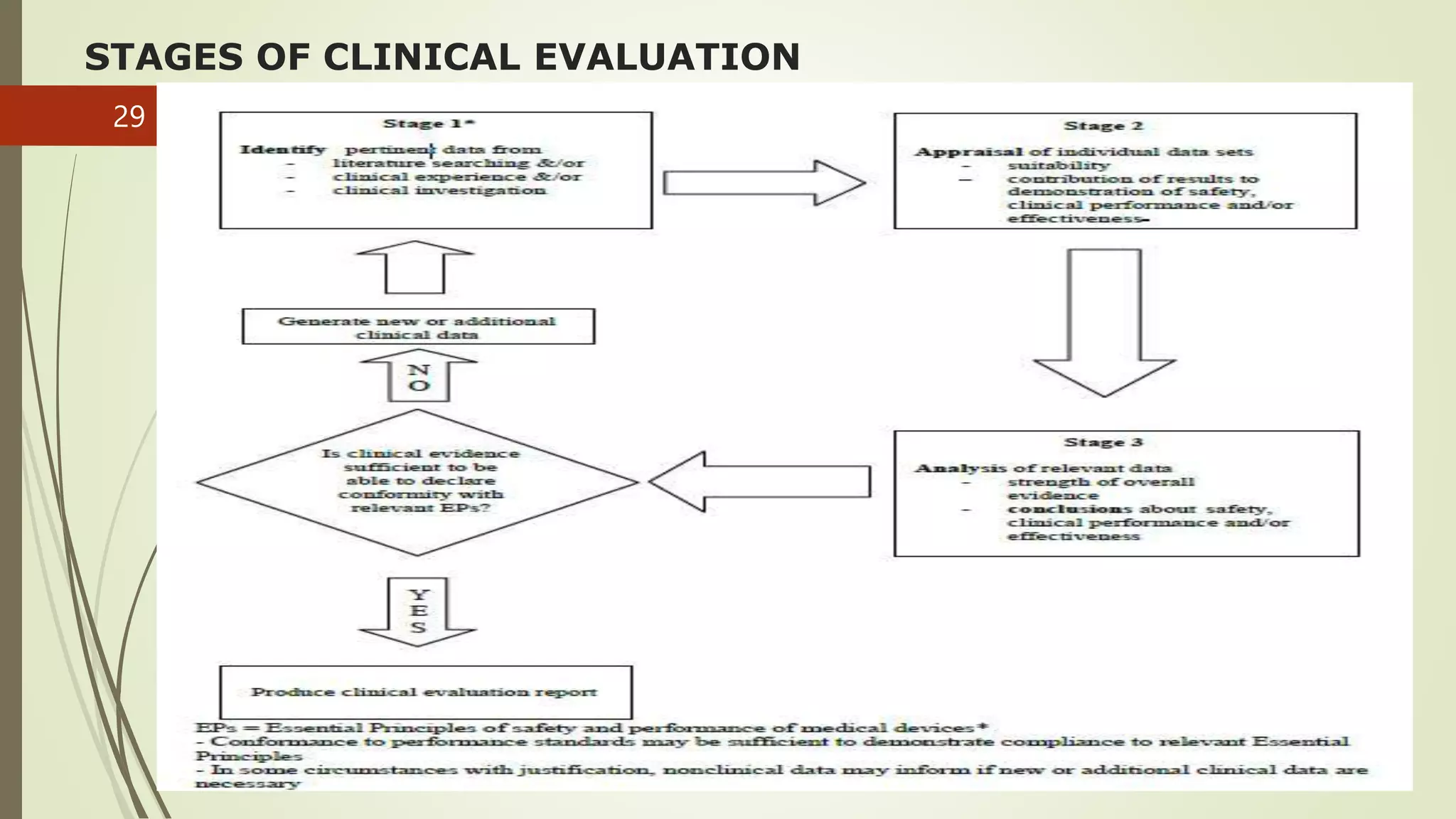
The document outlines the clinical investigation and evaluation processes for medical devices and in vitro diagnostics (IVDs), detailing definitions, regulatory frameworks, and classification systems across different jurisdictions. It emphasizes the importance of systematic investigations to assess safety and effectiveness, following established guidelines and ethical considerations. Furthermore, it stresses the need for comprehensive clinical evaluation procedures, incorporating both pre-market and post-market data to ensure compliance with safety and performance standards.





![Definitions
IVDS,
Definition: In vitro diagnostic products are those reagents,
instruments, and systems intended for use in diagnosis of disease or
other conditions, including a determination of the state of health, in
order to cure, mitigate, treat, or prevent disease or its sequelae. Such
products are intended for use in the collection, preparation, and
examination of specimens taken from the human body. [21 CFR 809.3]
IVDs are devices as defined in section 201(h) of the Federal Food,
Drug, and Cosmetic Act, and may also be biological products subject to
section 351 of the Public Health Service Act. Like other medical
devices, IVDs are subject to premarket and post market controls. IVDs
are generally also subject to categorization under the Clinical
Laboratory Improvement Amendments (CLIA '88) of 1988.
6](https://image.slidesharecdn.com/clinicalinvestigationandevaluationofmedicaldevicesand-230117161600-fef779bb/75/CLINICAL-INVESTIGATION-AND-EVALUATION-OF-MEDICAL-DEVICES-AND-pptx-6-2048.jpg)





























































![Educo Life Science [gathering clinical evidence] [module 1]](https://cdn.slidesharecdn.com/ss_thumbnails/educo-gatheringclinicalevidence-module1-220128131137-thumbnail.jpg?width=640&height=640&fit=bounds)


































