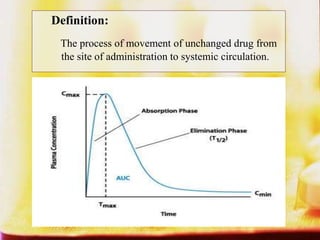
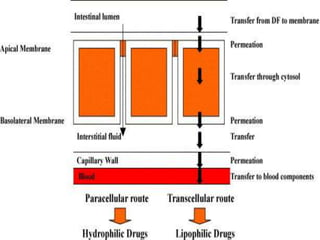
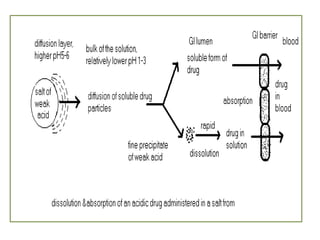
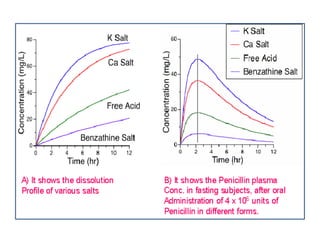
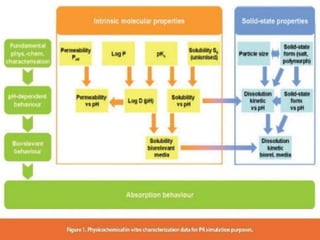
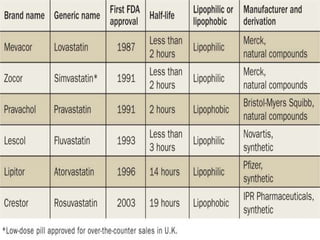
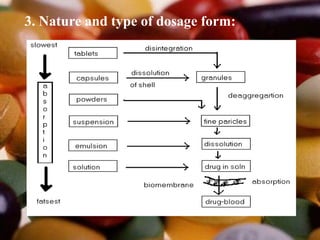
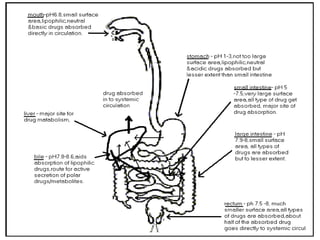
This document summarizes factors that affect drug absorption including pharmaceutical factors like drug solubility, particle size, polymorphism, and formulation factors as well as patient-related factors like gastric emptying time, gastrointestinal pH, and disease states. Some key pharmaceutical factors discussed are how smaller particle size increases surface area and dissolution rate, amorphous forms have higher solubility than crystalline forms, and solubility of different polymorphs and hydrates can impact absorption. The summary briefly outlines several formulation excipients and how they may influence drug absorption.