



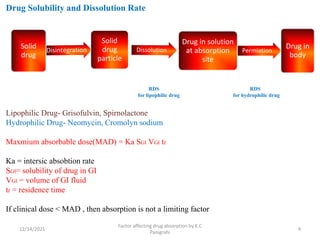
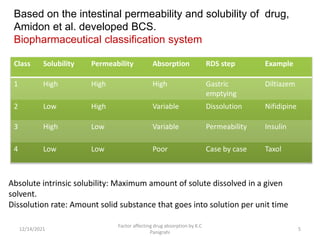

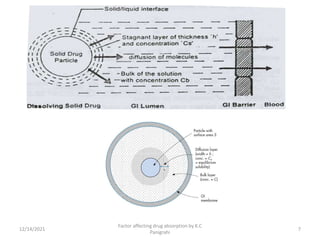



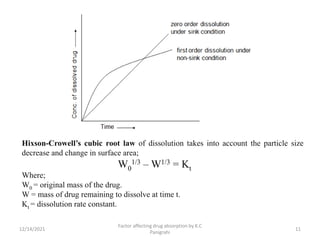

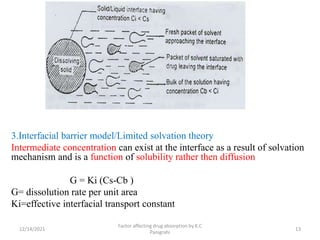


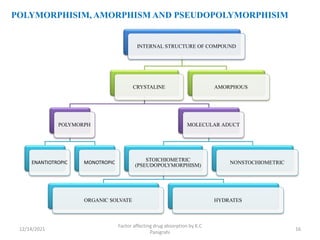






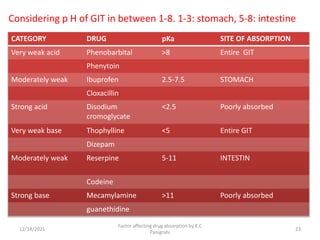






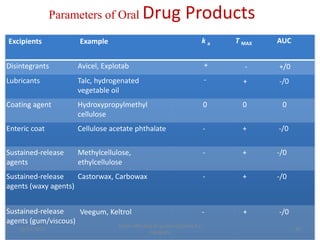
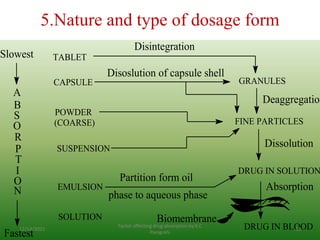


This document discusses factors that affect drug absorption in the gastrointestinal (GI) tract. It covers pharmaceutical factors like a drug's solubility, particle size, salt form, and polymorphism/amorphism, which can impact dissolution rate and absorption. It also discusses patient-related factors like age, GI pH, transit time, and disease status. Key pharmaceutical factors that influence drug absorption are solubility, dissolution rate, and factors that impact effective surface area like particle size.























































































