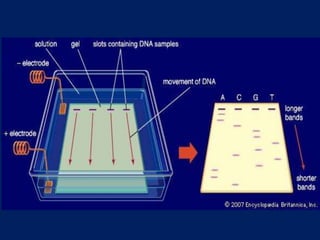
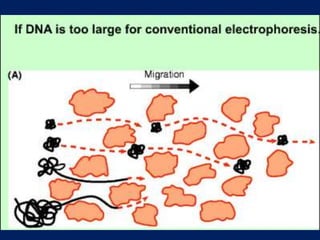
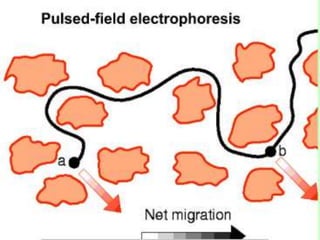
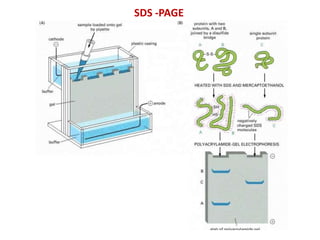
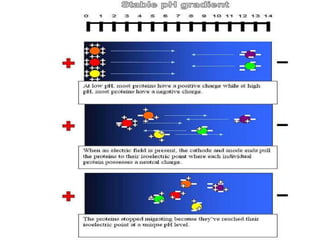
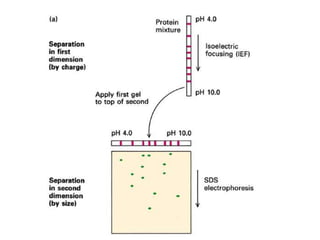
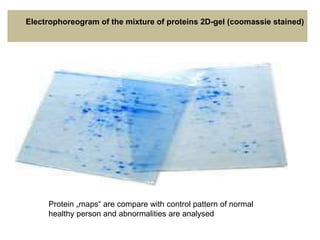
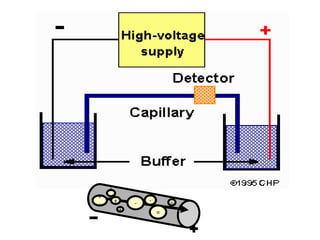
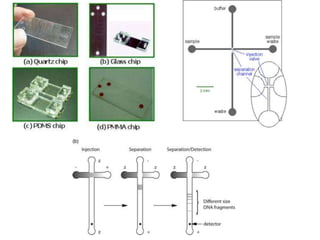
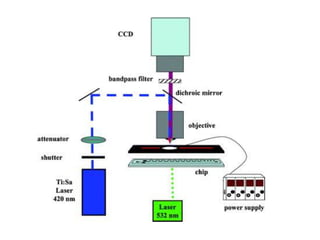
This document summarizes the principles and techniques of electrophoresis. Electrophoresis involves applying an electric field to move charged biomolecules like proteins and nucleic acids through a gel or liquid medium. It was first developed in the 1930s to study serum proteins. Factors like molecular charge, size, shape and buffer conditions determine electrophoretic mobility. Common applications include protein/nucleic acid analysis and purification. Techniques include agarose gel electrophoresis for DNA/RNA separation by size, and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to denature and linearly separate proteins by mass. Isoelectric focusing separates proteins based on their isoelectric point.