Downloaded 85 times






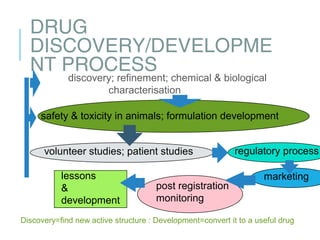

















The drug development process involves lengthy preclinical and clinical testing that can take over 12 years and cost $350 million. It begins with drug discovery followed by preclinical studies to test the drug's toxicity, pharmacokinetics, and efficacy in animals. If successful, an IND application is submitted to the FDA to begin human clinical trials. Clinical trials involve 4 phases to test the drug's safety and efficacy in humans. If phase 3 is successful, an NDA is submitted for FDA review and potential approval to market the new drug. Post-approval monitoring continues to ensure safety.























































































