The document introduces four students who are members of the team "Pakistan Pharma Career Door". It provides brief descriptions of each student, noting their educational achievements and strengths. Sameeta Malik is an energetic student engaged in scientific activities at Dow College of Pharmacy. Iffrah Naushad is a meritorious student with experience attending national events. Abira Khalid is an associate of Dow College of Pharmacy who is talented and one of the genius students. Qaisara Boota is one of the most active students who is highly courageous and ready to take on difficult tasks. The document was prepared by these four students from Dow College of Pharmacy.








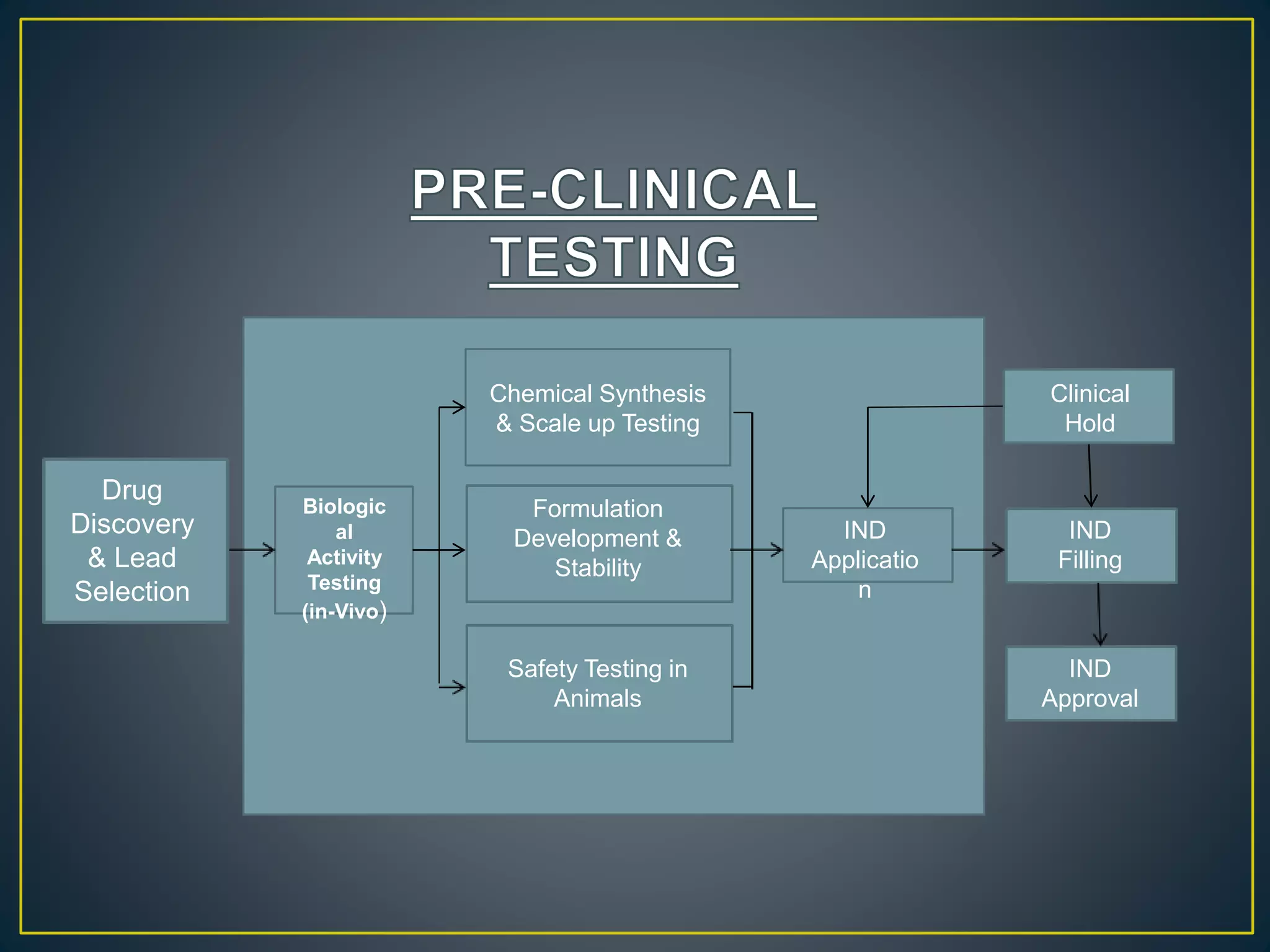
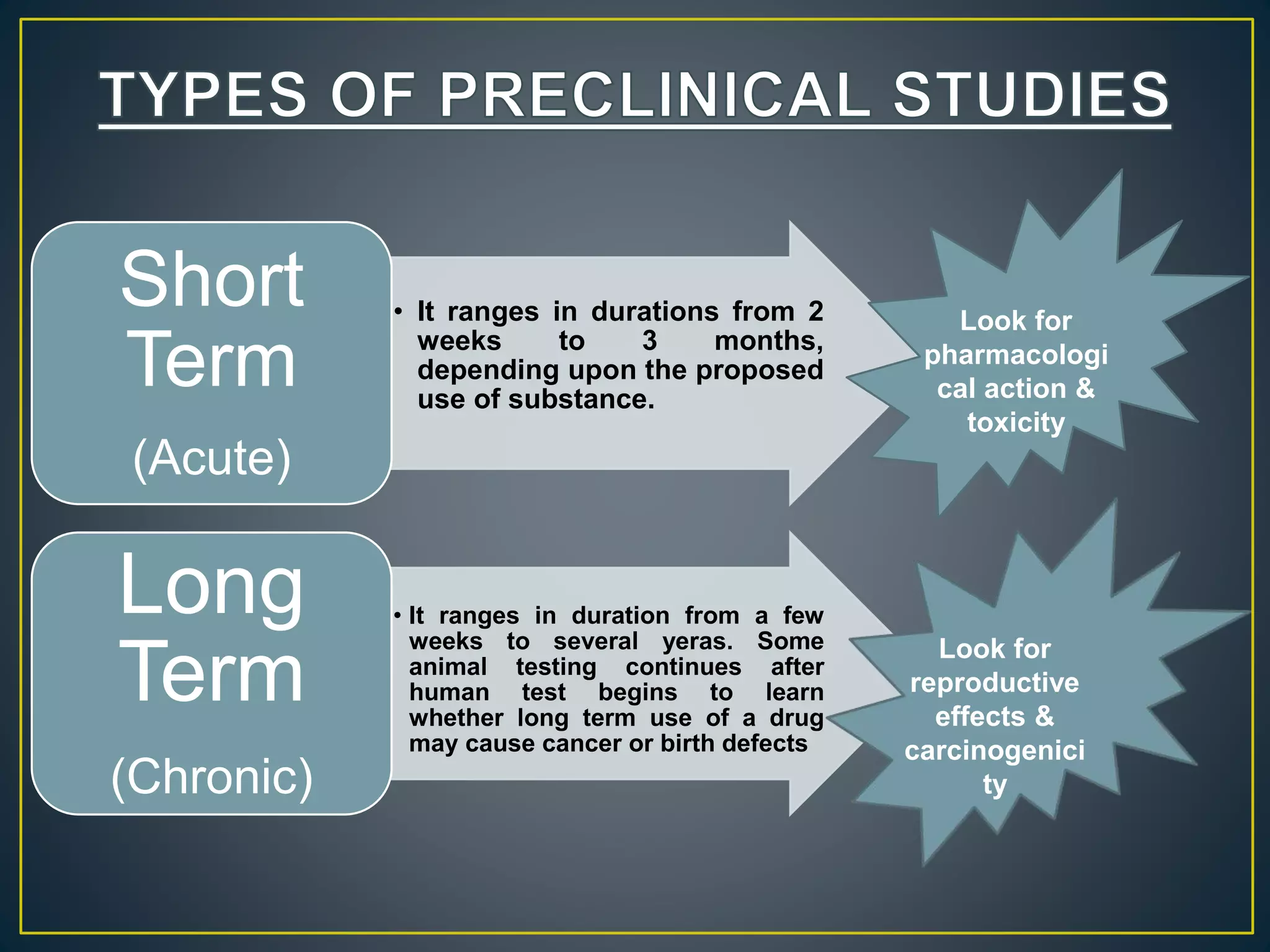
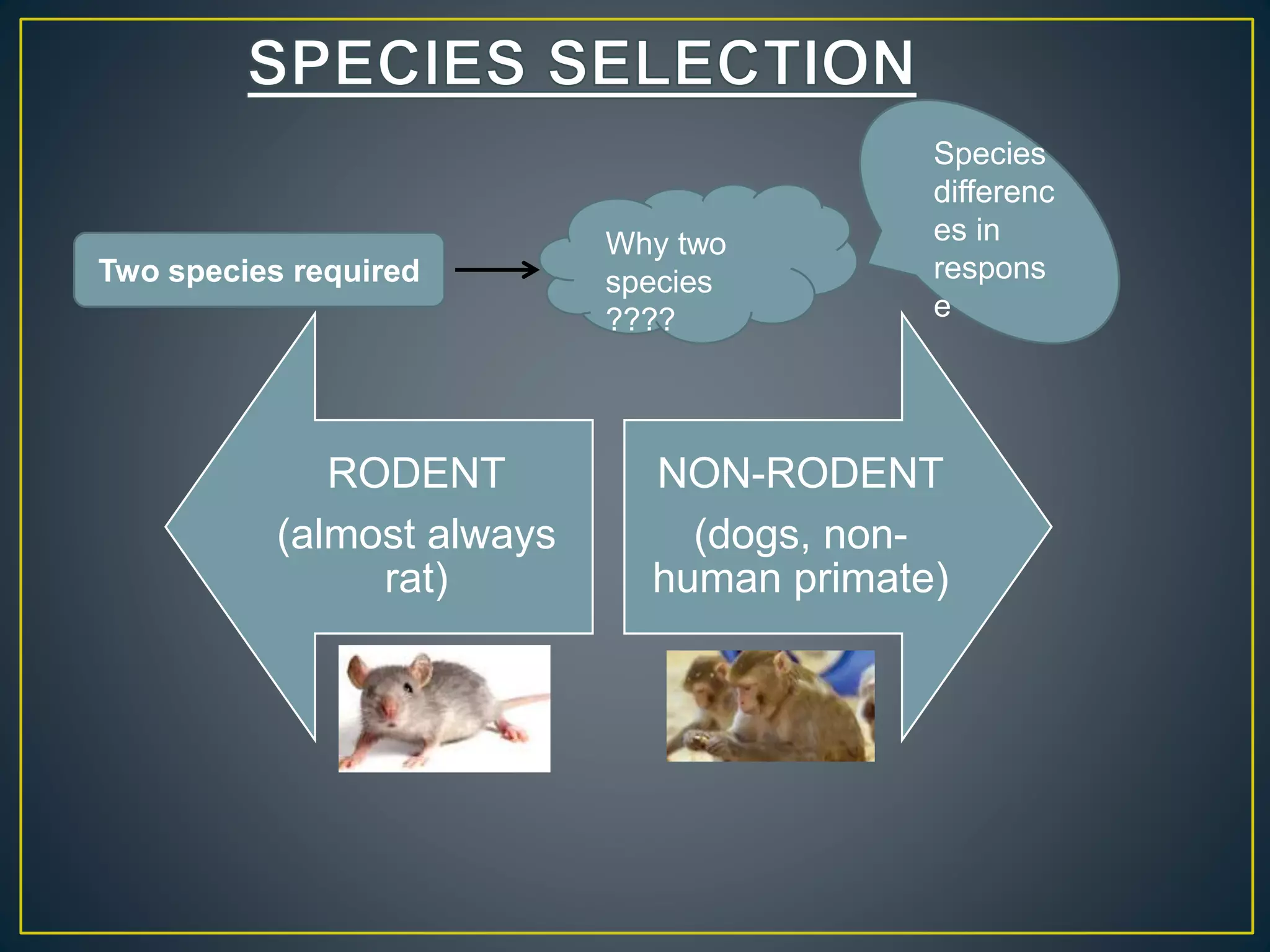


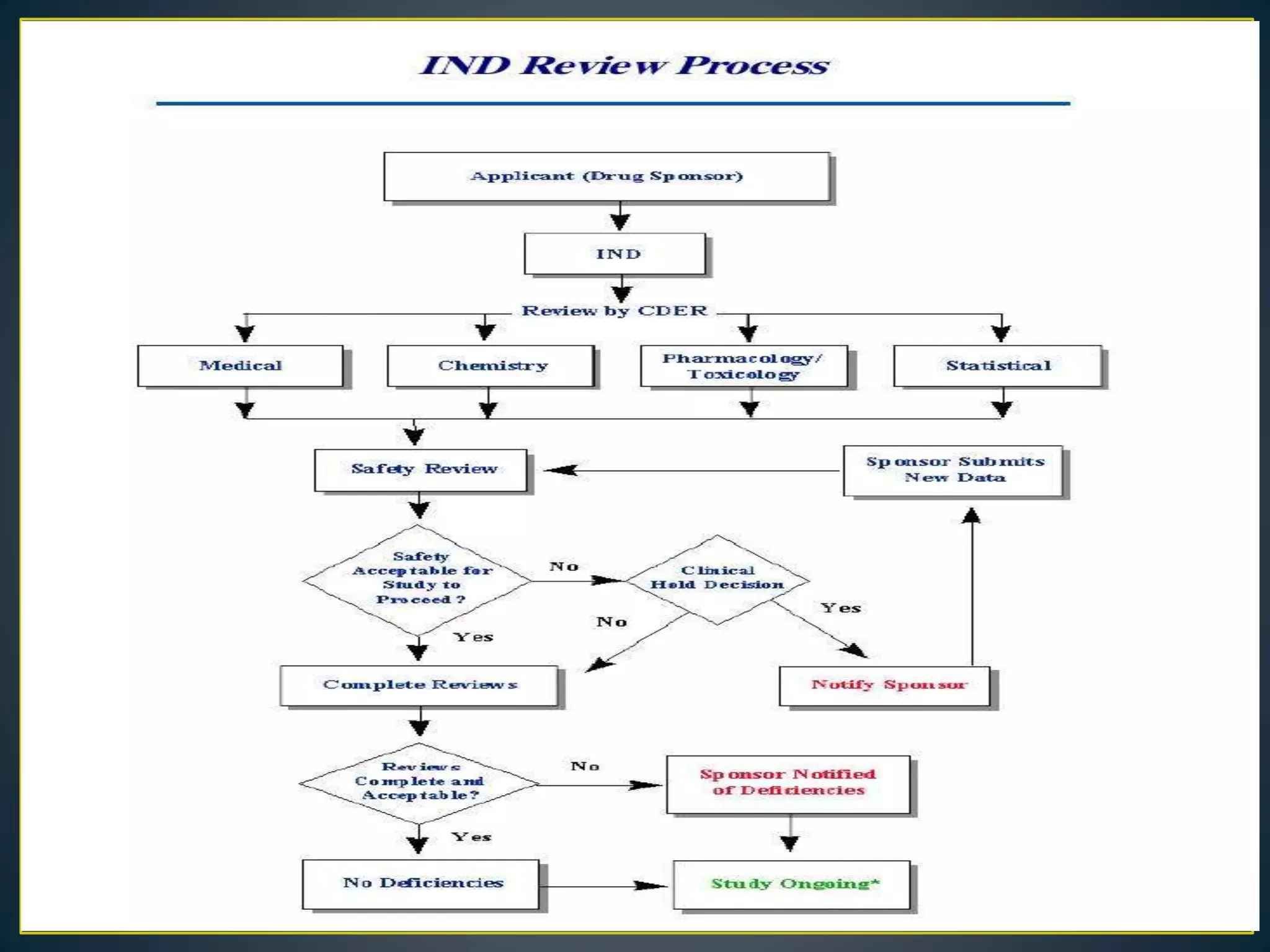



















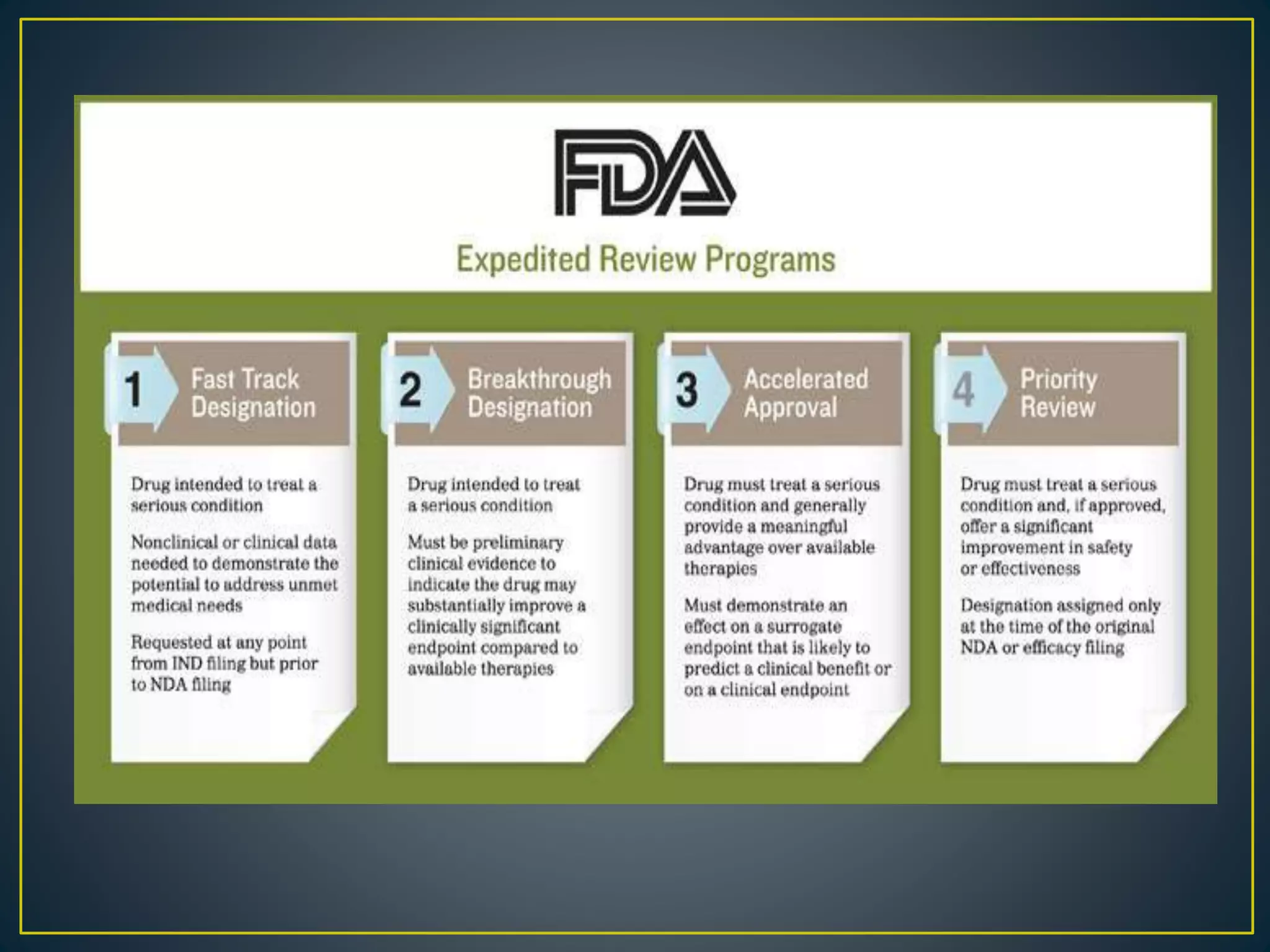





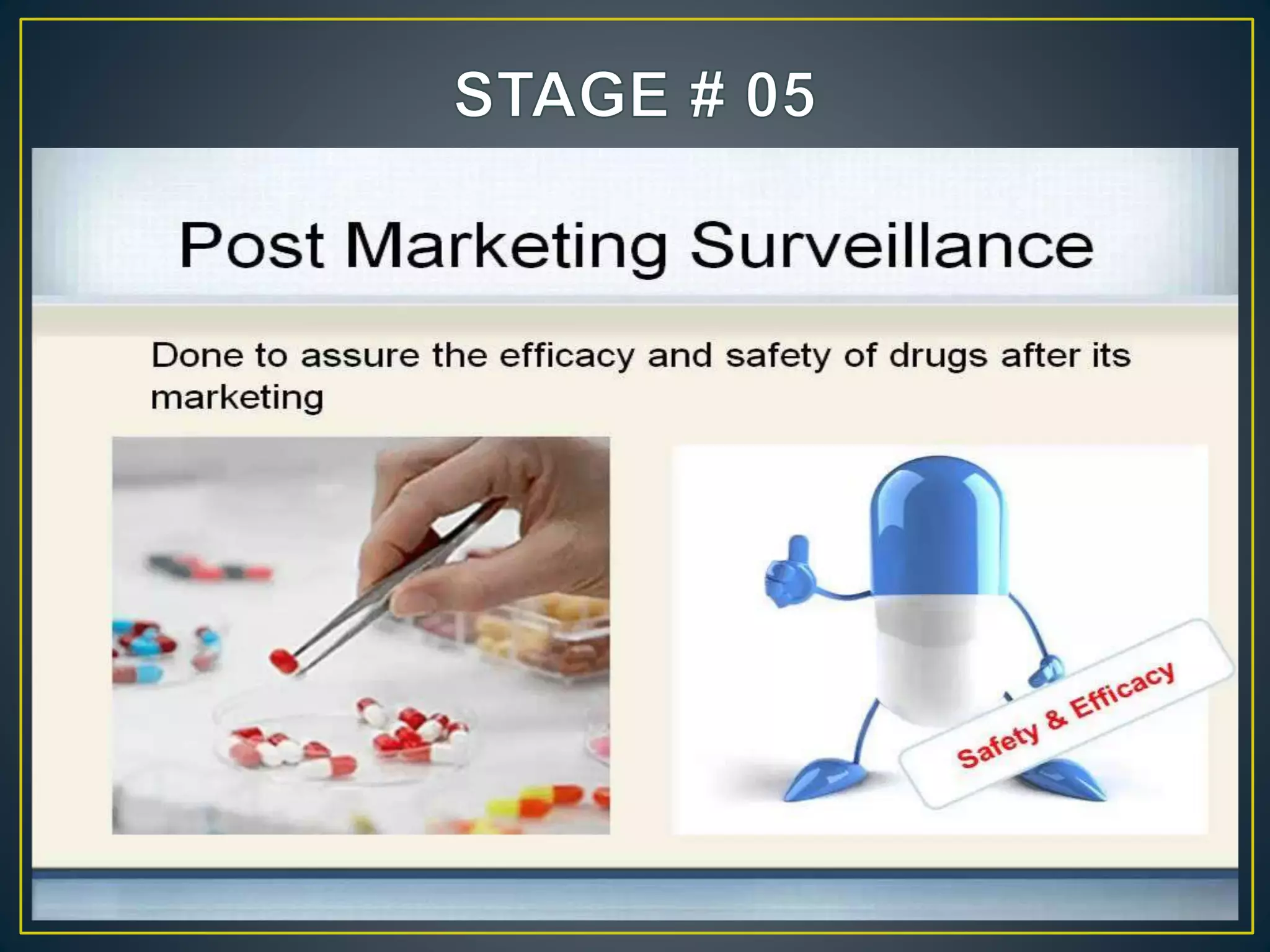


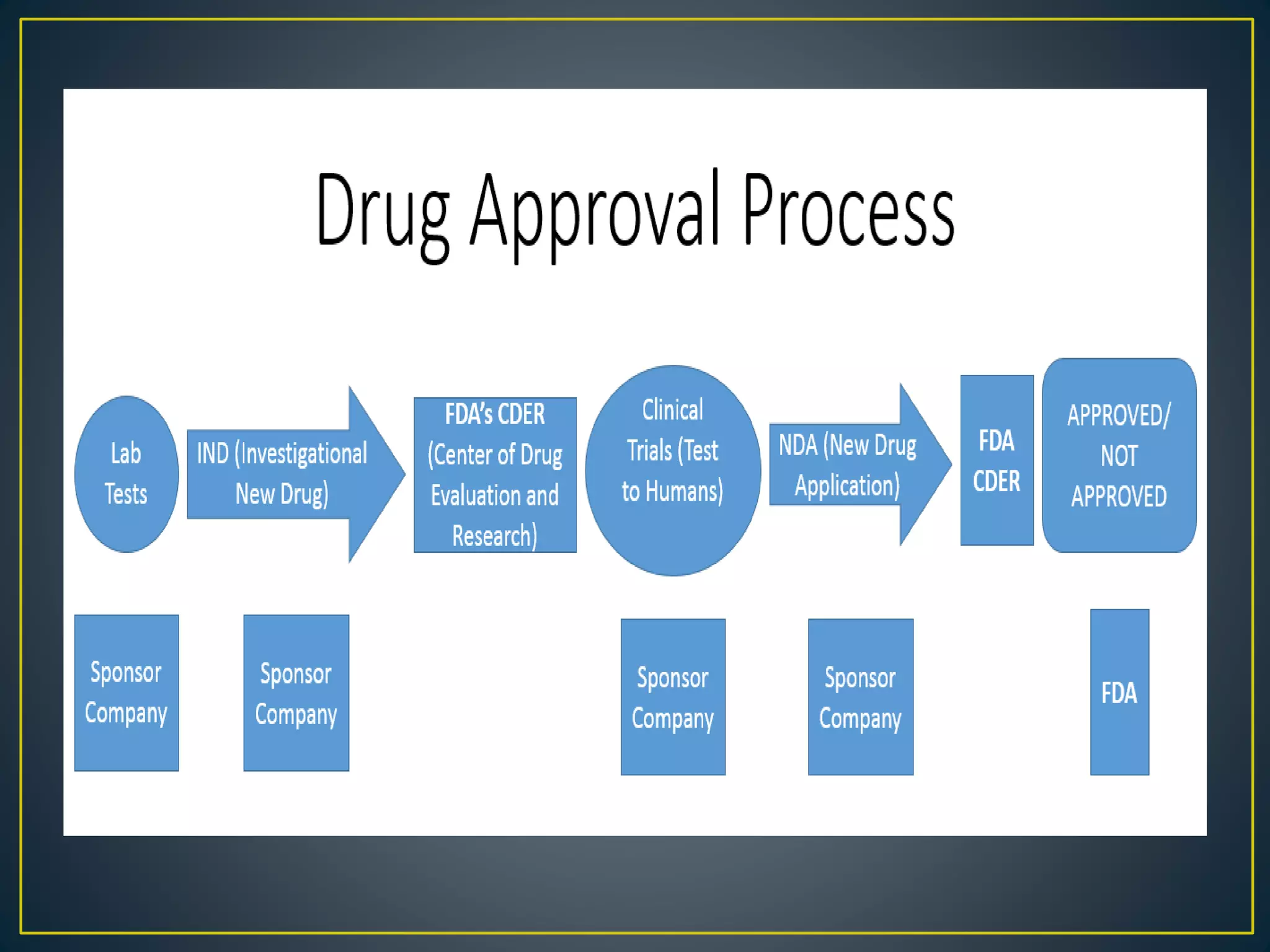


























![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)































































