Download as PDF, PPTX

















































![DOPA
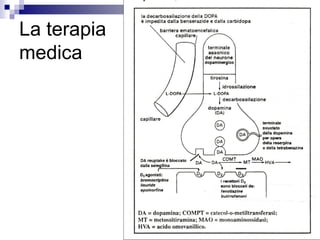
Levodopa is clearly the
most potent drug for
controlling PD symptoms,
particularly those related
to bradykinesia [Jankovic,
2002].
The therapeutic benefits of
levodopa have been
markedly enhanced by
the addition of carbidopa,
a peripheral dopa
decarboxylase inhibitor.
Absence of morning
kick…](https://image.slidesharecdn.com/disordinidelmovimento-150402123336-conversion-gate01/85/Disordini-del-movimento-v-61-320.jpg)










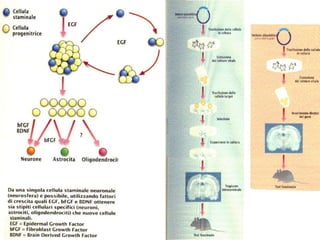


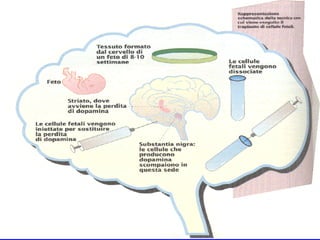
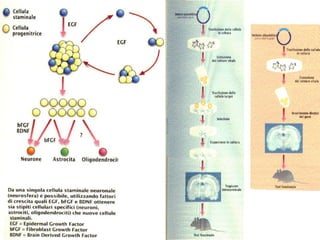













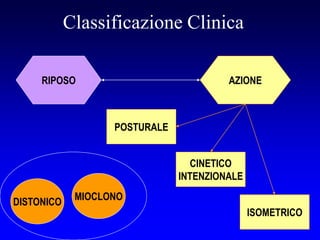
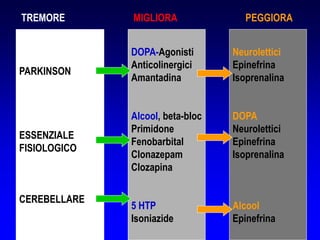

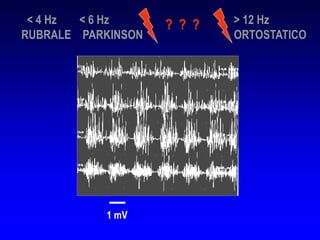










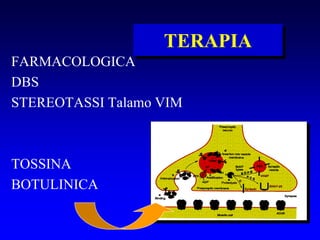












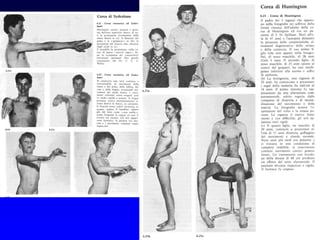







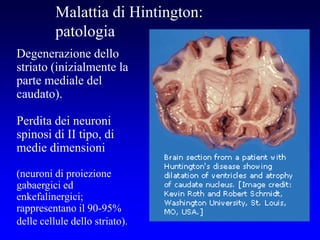









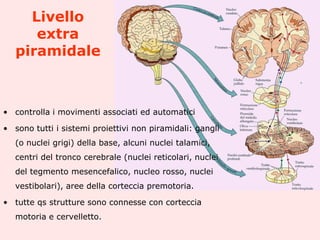
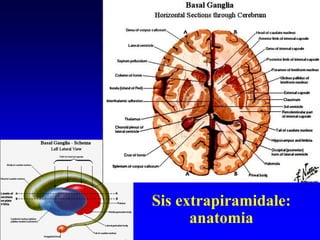
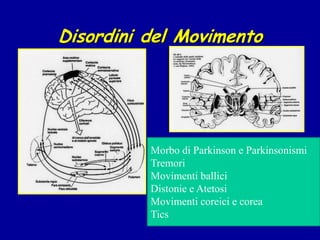
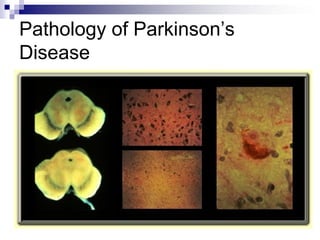
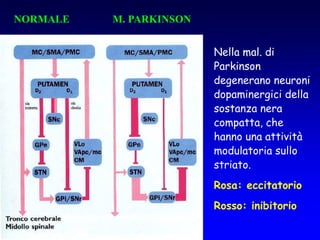
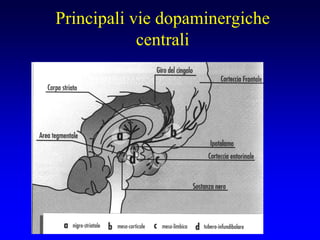
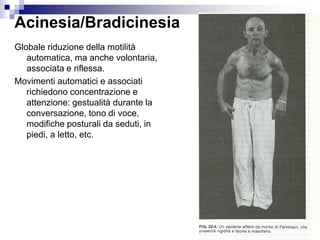
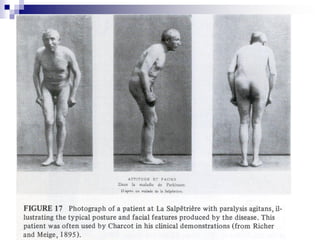
Il documento discute i disordini del movimento, concentrandosi sulla malattia di Parkinson, una patologia neurologica caratterizzata da acinesia, rigidità e tremore, causata da una degenerazione dei neuroni dopaminergici nel sistema extrapiramidale. Esamina anche la fisiopatologia, la diagnosi, i sintomi clinici e le opzioni terapeutiche, sia farmacologiche che chirurgiche, per la gestione della malattia. Viene evidenziata l'importanza di un approccio multidisciplinare per migliorare la qualità della vita dei pazienti affetti da questa condizione.





















![2009 Convegno Malattie Rare Palmucci [22 01]](https://cdn.slidesharecdn.com/ss_thumbnails/2009convegnomalattierarepalmucci2201-090330150816-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)












































