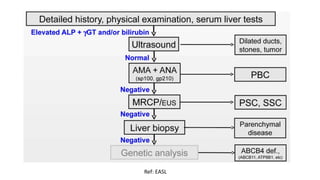
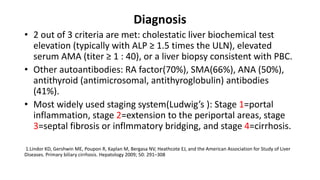
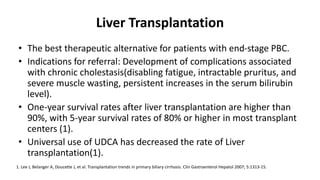
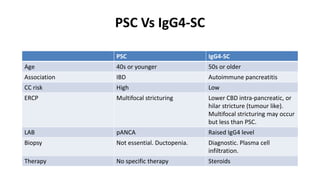
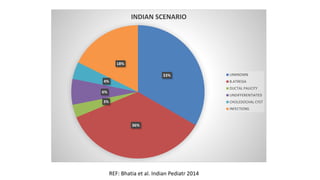
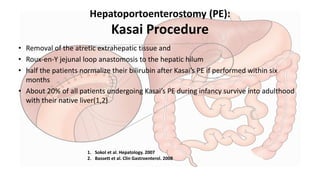
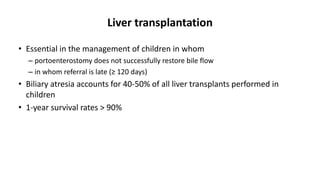
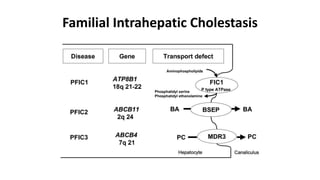
This document provides an overview of the management of cholestatic syndromes. It begins by defining cholestasis and describing the cell biology of cholangiocytes. It then classifies cholestasis as either intrahepatic or extrahepatic. Several specific cholestatic diseases are discussed in detail, including primary biliary cirrhosis, primary sclerosing cholangitis, IgG4-associated cholangitis, and intrahepatic cholestasis of pregnancy. Diagnostic tests and treatment approaches for each condition are summarized.