Department of Chemistry, MIT, Manipal
by
DR. SANTOSH L GAONKAR
Professor, Department of Chemistry,
Manipal Institute of Technology
Manipal Academy of Higher Education , Manipal
Catalysis
1
2.
Introduction to Catalysis
•Catalysis is the acceleration of a chemical
reaction by a substance called a catalyst,
which is not consumed in the reaction.
• Catalysts enhance reaction rates without
altering equilibrium positions.
• They may be solid, liquid, or gas, and often
work via complex formation with reactants.
3.
Introduction to Catalysis
KeyFeatures of Catalysis
• Lowers activation energy of a reaction.
• Does not alter the overall thermodynamics
(i.e., the equilibrium remains unchanged).
• Catalyst remains unchanged after the
reaction.
• Enables selective and efficient
transformations.
4.
Characteristics
Catalysts play acrucial role in increasing the rate
of chemical reactions
• Not consumed in the reaction
• Highly specific
• Small amount is sufficient
• Poisoning (deactivation by impurities)
• Require promotors or inhibitors
5.
Types of Catalysis
•Homogeneous Catalysis: Catalyst and reactants in the same
phase (e.g., Co carbonyl in hydroformylation).
• Heterogeneous Catalysis: Catalyst and reactants in different
phases (e.g., Ni in edible oil hydrogenation).
• Electrocatalysis: Electron transfer at electrodes (e.g., chlor-
alkali electrolysis).
• Photocatalysis: Light-driven reactions (e.g., TiO₂ degradation of
organics).
• Biocatalysis: Enzyme/microorganism-catalyzed reactions.
• Environmental Catalysis: Emission control (e.g., NOx
reduction).
• Green Catalysis: Sustainable and selective catalytic processes.
6.
Catalysis as aScientific Discipline
• Catalysis research spans mechanisms, catalyst
design, and industrial applications.
• Key journals: Journal of Catalysis, Applied
Catalysis, Catalysis Today, ACS Catalysis etc.
• Institutes and companies worldwide are
dedicated to catalytic research and
innovation.
7.
History of Catalysis
•1835: Catalysis defined by Berzelius.
• 1895: Ostwald defines catalysis; wins Nobel in
1909.
• 1912: Sabatier awarded Nobel for
hydrogenation research.
Catalytic processes like fermentation and ether
formation date back centuries.
8.
Industrial Importance
• Over80% of industrial chemical processes use
catalysts.
• Catalysis is crucial in petrochemical,
pharmaceutical, polymer, and environmental
sectors.
• More than 15 global companies specialize in
catalyst production.
9.
Theoretical Aspects
• Catalysisalters reaction rates, not
thermodynamics.
• Reaction conditions vary widely (78K–1500K,
10⁻⁹–100 MPa).
• Reactions may involve gases, solvents, light,
radiation, or electricity.
• Site-time yields range from 10⁻⁵ to 10⁹ s⁻¹.
• No universal theory exists; general principles
and specific concepts are used.
10.
Why Mechanisms Matter
Understandingcatalytic mechanisms allows scientists to:
•Design more efficient catalysts (higher activity/selectivity)
•Develop new reactions for cleaner or more economical
processes
•Improve catalyst life by addressing deactivation pathways
•Tailor reaction conditions for industrial-scale synthesis
11.
Modifiers and Promoters
•Promoters increase catalyst activity.
• Poisons reduce activity or selectivity.
• Additives can enhance selectivity or prevent
degradation.
• Example: K₂O and Al₂O₃ promote Fe-based
ammonia synthesis catalysts.
12.
Catalyst Deactivation Mechanisms
•Sintering: Particle agglomeration at high
temperature (e.g., Ni catalysts).
• Poisoning: Strong adsorption of impurities like
S or Pb blocks active sites.
• Fouling: Deposition of coke or dust blocks
pores or surface sites.
13.
Sintering in Catalysis
•Sintering is the agglomeration of catalyst particles
at high temperature.
• Reduces surface area and number of active sites.
• Types: Ostwald ripening, particle migration and
coalescence.
• Common in Ni, Pt, and Rh catalysts during
reforming or high-temp processes.
• Prevention: Use of stabilizers (e.g., Al2O3), lower
regeneration temperatures.
14.
What Happens DuringSintering?
•Catalyst particles merge or grow, reducing the number of
exposed active sites.
•This typically occurs at elevated temperatures during
operation or regeneration.
•It’s especially problematic in metallic catalysts (e.g., Ni, Pt,
Rh).
Types of Sintering:
1.Ostwald Ripening:
1. Smaller particles dissolve and redeposit onto larger
particles via vapor or surface diffusion.
2. Driven by the thermodynamic preference for lower surface
energy.
2.Particle Migration and Coalescence:
1. Whole particles move and fuse into larger ones on the
support surface.
15.
Consequences:
•Reduced surface area→ fewer active sites.
•Lower activity and longer startup times.
•Irreversible in most cases → requires catalyst
replacement.
Common in:
•High-temperature reactions: e.g., steam reforming,
ammonia synthesis.
•Poorly supported or unanchored metal catalysts.
•Systems with metal catalysts on low-surface-area supports.
Prevention Strategies:
•Use of thermal stabilizers or promoters (e.g., Al₂O₃, MgO).
•Strong metal-support interaction (SMSI) to anchor particles.
•Operating under lower regeneration temperatures.
Example:
•Nickel catalysts used in methane reforming often sinter above
700°C, reducing hydrogen yield over time.
16.
Poisoning in Catalysis
•Poisoning is caused by strong adsorption of
impurities on active sites.
• Leads to irreversible or reversible deactivation.
• Common poisons: Sulfur, lead, chlorine,
phosphorus.
• Examples: Sulfur poisoning of Ni in steam
reforming.
• Prevention: Feedstock purification, poison-
resistant catalysts, guard beds.
17.
What Happens DuringPoisoning?
•A poison molecule binds irreversibly or very strongly to the active
site.
•This blocks the access of reactants to those sites.
•The overall catalytic activity drops significantly.
18.
Fouling in Catalysis
•Fouling is physical blockage of catalyst pores or
surfaces by unwanted deposits.
• Common foulants: Coke, ash, tar, heavy metals, dust.
• Reduces diffusion, increases pressure drop, lowers
activity.
• Examples: Coke fouling in catalytic craking, tar
fouling in biomass gasification.
• Prevention: Feed pretreatment, guard beds, periodic
regeneration.
19.
Mechanism:
1.Reactants pass overthe catalyst.
2.Side reactions or impurities produce non-volatile by-
products.
3.These by-products stick to the catalyst surface or
accumulate in pores.
4.Active sites become physically blocked, leading to
reduced catalytic activity.
Types of Catalysis
•Homogeneous catalysis is a type of catalysis where the catalyst
and the reactants exist in the same physical phase, most
commonly in the liquid phase.
• This setup allows for uniform mixing and molecular-level
interactions, leading to well-defined reaction mechanisms.
Key Features:
• Same phase: Typically, both the catalyst and reactants are
dissolved in a solvent (usually a liquid).
• Uniform reaction environment: Since all species are in the same
phase, the catalyst can interact directly with reactants throughout
the solution.
• Well-understood mechanisms: Molecular-level interactions allow
for precise control and study of the catalytic process.
Homogeneous catalysis
22.
Examples:
1.Hydroformylation of alkenes
1.Catalyst: Cobalt or rhodium carbonyl complexes
2. Reaction: Converts alkenes into aldehydes using CO and H₂.
2.Acid-catalyzed esterification
1. Catalyst: H₂SO₄ (sulfuric acid)
2. Reaction: Alcohol + Acid → Ester + Water
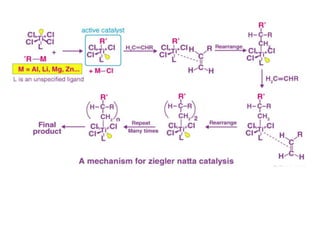
3.Zeigler-Natta polymerization (early versions)
1. Catalyst: TiCl₄ and Al(C₂H₅)₃ in solution for producing
polyethylene and polypropylene.
23.
Advantages:
•High selectivity andactivity.
•Reaction mechanism is easier to study and control.
•Catalyst is well-dispersed, ensuring efficient contact
with reactants.
Disadvantages:
•Difficult catalyst recovery after the reaction.
•Thermal instability in some cases.
•Often requires special separation techniques (e.g.,
distillation, solvent extraction).
24.
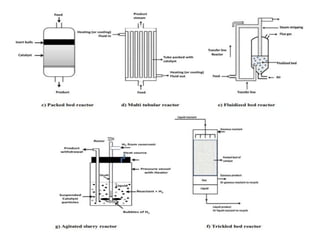
Heterogeneous Catalysis:
Heterogeneous catalysisoccurs when the catalyst and
reactants are in different physical phases, typically a solid
catalyst with liquid or gas phase reactants. This is the most
widely used type of catalysis in industrial applications.
Key Features:
•Phase difference: Usually, the catalyst is a solid, while
reactants are gases or liquids.
•Reaction occurs at the catalyst surface, making surface
area and active site accessibility critical.
•Common in large-scale processes due to ease of catalyst
separation and reuse.
25.
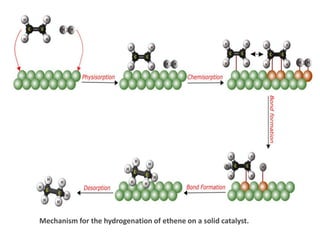
Mechanistic Note:
Steps typicallyinclude:
1. Adsorption of reactants onto the surface
2. Reaction on the surface
3. Desorption of products
Adsorption
Molecule in thegas or solution phase binds to a solid surface. There are two
forms of adsorption: physisorption and chemisorption.
•Physisorption:
In physisorption, the molecule adheres to the surface through the action of
van der Waals forces. Generally, physisorption occurs before chemisorption
as an intermediate energy state.
•Chemisorption:
is a type of adsorption in which the surface and the adsorbate undergo a
chemical interaction. As a result, new chemical bonds are formed at the
adsorbent surface.
Surface Reactions
Adsorption sites are not usually active catalyst sites. Therefore, the reactant
molecules must move over the surface to reach an active site. Then the
reactant molecules will react at the active site to create product molecules.
In fact, this product formation takes a more energy-efficient route via
catalytic intermediates.
Desorption
It is the reverse of adsorption. That is, the product molecules will separate
from the catalyst surface during desorption. Finally, the products will diffuse
out from the solid surface into the fluid phase.
28.
Typical heterogeneous catalystsare inorganic solids such
as metals, oxides, sulfides, and metal salts, but they may
also be organic materials such as organic hydroperoxides,
ion exchangers, and enzymes
Example:
Hydrogenation nitrocompounds Pt/Pd/Ni catlysts
Hydrogenation of edible oils
•Catalyst: Nickel (Ni) solid on a porous support (e.g.,
kieselguhr)
•Reactants: Liquid vegetable oil + hydrogen gas
•Product: Saturated fats
29.
Disadvantages:
•Mass transfer limitations(reactants must reach catalyst
surface).
•Active site heterogeneity makes the reaction
mechanism complex.
•Possible deactivation due to sintering, poisoning, or
fouling.
Advantages:
•Easy catalyst recovery and reuse.
•Highly stable under harsh industrial conditions.
•Can be used in continuous reactors (e.g., fixed-bed or
fluidized-bed).
30.
Electrocatalysis is aspecial case of heterogeneous
catalysis involving oxidation or reduction by electron
transfer. Examples include the use of catalytically active
electrodes in electrolysis processes such as chlor-alkali
electrolysis and in fuel cells.
31.
In photocatalysis
Photocatalysis involvesthe acceleration of chemical
reactions using light and a photocatalyst, usually a
semiconductor like TiO₂. It is widely used in environmental,
energy, and chemical applications.
Light is absorbed by the catalyst or a reactant during the
reaction.
This can take place in a homogeneous or heterogeneous
system.
example is the utilization of semiconductor catalysts
(titanium, zinc, and iron oxides) for photochemical
degradation of organic substances,
32.
Air Purification
•Photocatalyst: TiO₂(often coated on surfaces)
•Application: Removal of NOx, SOx, VOCs, and other
pollutants from air
•Industries: HVAC systems, tunnel air cleaning, self-cleaning
walls
Water & Wastewater Treatment
•Photocatalyst: TiO₂, ZnO
•Application: Degradation of organic dyes, pesticides,
pharmaceuticals, and pathogens in contaminated water
•Industries: Textile, pharmaceutical, municipal
wastewater plants
33.
Self-Cleaning Surfaces
•Photocatalyst: TiO₂-coatedglass, ceramics, paint
•How it works: UV light activates TiO₂, which degrades organic
matter and repels water (superhydrophilic)
•Industries: Construction, automotive (e.g., Pilkington Activ
self-cleaning glass)
Hydrogen Production (Photocatalytic Water Splitting)
•Photocatalyst: Modified TiO₂, ZnO, CdS, g-C₃N₄
•Application: Splitting water into H₂ and O₂ using solar energy
•Industries: Emerging in green hydrogen and renewable energy
sectors
34.
CO₂ Reduction /Fuel Production
•Photocatalyst: Cu-doped TiO₂, metal–organic frameworks
(MOFs)
•Application: Convert CO₂ and water into methanol, methane,
or formic acid using sunlight
•Goal: Reduce greenhouse gases and produce solar fuels
Antibacterial and Antiviral Coatings
•Photocatalyst: TiO₂ (sometimes doped with Ag or Cu)
•Application: Surfaces that kill bacteria or viruses under light
(including visible or UV)
•Industries: Hospitals, public transport, consumer goods
(COVID-19 boosted demand)
35.
In biocatalysis,
enzymes ormicroorganisms catalyze various biochemical
reactions. The catalysts can be immobilized on various
carriers such as porous glass, SiO2, and organic polymers.
Prominent examples of biochemical reactions are
isomerization of glucose to fructose, important in the
production of soft drinks, by using enzymes such as
glucoamylase immobilized on SiO2,
Conversion of acrylonitrile to acrylamide by cells of
corynebacteria entrapped in a polyacrylamide gel.
36.
The main aimof environmental catalysis is environmental
protection.
Examples are the reduction of NOx in stack gases with
NH3 on V2O5–TiO2 catalysts and the removal of NOx,
CO, and hydrocarbons from automobile exhaust gases by
using the so-called three-way catalyst consisting of Rh–
Pt– CeO2–Al2O3 deposited on ceramic honeycombs.
37.
The term greencatalytic processes used frequently in
recent years, implying
• chemical processes may be made environmentally
benign by taking advantage of the possible high yields
and selectivity for the target products,
• little or no unwanted side products and also often high
energy efficiency.
The basic chemical principles of catalysis consist in the
coordination of reactant molecules to central atoms, the
ligands of which may be molecular species
(homogeneous and biocatalysis)
or neighboring atoms at the surface of the solid matrix
(heterogeneous catalysis).
38.
Solid catalysts
Catalyst components
Asolid catalyst consists of mainly three components :
1. Catalytic agent
2. Support /carrier
3. Promoters and Inhibitors
Catalytic agent:
These are the catalytically active component in the
catalyst.
These components generate the active sites that
participate in the chemical reaction.
Activity of any catalyst is proportional to the concentration
of these active sites.
39.
Catalytic agents maybe broadly divided in the following categories:
i. Metallic conductors ( e.g Fe, Pt, Ag, etc.)
ii. Semiconductors (e.g. NiO, ZnO,etc.)
iii. Insulators (e.g. Al2O3, SiO2,MgO etc.)
Metallic conductors:
The metals that have strong electronic interaction with the
adsorbates are included in this category.
The metals are used in various catalytic reactions such as methanol
synthesis, oxidation , hydrogenation and dehydrogenation
processes.
Examples of metal catalysts :
Cu for water gas shift reaction and methanol synthesis ;
Ag for oxidation of ethylene to ethylene oxide,
Au, Ag and iron-molybdenum for oxidation of methanol to formaldehyde;
Fe for ammonia synthesis;
Pd and Pt for hydrogenation of olefins, dienes, aniline or nitriles as well as
dehydrogenation of alkanes, alcohols, cyclohexanes, cyclohexanols etc
40.
Semiconductors :
The oxidesand sulfides of transition metals that have
catalytic activity are included in this category.
Similar to conducting metals, they are also capable of
electronic interaction with adsorbed species and catalyze
the same type of reactions.
Common transition metal oxides and sulfides such as CuO,
AgO, NiO CoO, Fe2O3 , MnO, Cr2O3, FeS, V2O5 show
conductivity.
These materials participate in catalytic reactions and
reaction occurs through acceptation or donation of
electrons between the reactant material and catalysts
Examples:
CuO for oxidation of nitric oxides, NiO for dehydrogenation of alkanes, MnO2 for
oxidation of alcohols, and V2O5 for oxidation of hydrocarbons.
41.
Insulators :
Catalytic functionsof insulators are different from that of
conductor and semi conductor materials.
Insulators have large values of band gap energy and very
low concentration of impurity levels. The electrons remain
localized in valence bonds and redox type reactions
involving electronic interaction as observed for metal or
semiconductor catalysts does not occur.
However, insulators have sites that generate protons,
thereby, promote carbonium ion-based reactions such as
cracking, isomerization or polymerization.
Al2O3, SiO2, SiO2-Al2O3, zeolites, MgO, CaO, MgAl2O4,
SiO-MgO are few examples of the insulators used as
catalysts.
42.
Support or carrier
Inheterogeneous catalysis, a support or carrier is an inert or semi-
inert solid material on which a catalytically active phase (usually a
metal or metal oxide) is dispersed.
Though the support itself is not usually catalytically active, it plays a
crucial role in the performance of the catalyst system.
43.
Role of CatalystSupports
• Supports provide surface area and mechanical
strength.
• Influence dispersion and thermal stability of
active phase.
• Often chemically inert but can modify catalyst
performance.
44.
Functions of aSupport in Catalysis:
Dispersing the Active Component:
•Supports increase the surface area over which the active species
(like Pt, Pd, Ni, Cu, etc.) are dispersed.
•This improves exposure to reactants and enhances catalytic
efficiency.
Mechanical Stability:
•Supports provide physical strength and thermal durability, especially
important in high-temperature reactions (e.g., steam reforming,
oxidation).
45.
Functions of aSupport in Catalysis:
Control of Pore Structure and Prevention of Sintering:
•Porous supports offer defined pore sizes (mesoporous,
microporous), which control mass transport and selectivity (shape-
selective catalysis in zeolites).
• Supports prevent agglomeration of catalysts
Chemical Interactions (Promoters or Modifiers):
•Some supports modify the electronic or geometric properties of the
active phase, improving activity, selectivity, or resistance to poisoning
(e.g., strong metal–support interaction or SMSI effect in TiO₂-
supported catalysts).
46.
Common Catalyst Supports:
Pt/Al₂O₃catalyst in reforming:
•Pt is the active metal.
•Al₂O₃ disperses Pt finely and withstands high temperatures.
47.
Promoters :
• Substancesadded during preparation of catalysts that
improve the activity or selectivity or stabilize the
catalytic agents.
• The promoter is present in a small amount and by itself
has little or no activity.
• Promoters are termed as physical or chemical
promoter depending on the manner they improve the
catalyst performance.
48.
Promoters :
• Theadditives that maintain physical integrity of the
support and/or deposited catalytic agents are termed
as physical promoters.
• For example, addition of small quantities of alumina to
an iron catalyst employed in ammonia synthesis
prevents sintering of the iron crystallites. Thus, for this
catalyst, alumina is a physical promoter.
• The addition of K2O to the same catalyst increases the
intrinsic activity of the iron crystallites and therefore
acts as a chemical promoter.
The promoter can be added during catalyst preparation or
during reaction.
49.
Negative promoters orinhibitors:
• Inhibitors act opposite to promoters.
• When added in small amounts, these can reduce
catalyst activity, selectivity or stability.
• Inhibitor is particularly useful for reducing the activity
of a catalyst for undesirable side reactions.
• In oxidation of ethylene, ethylene dichloride is added
to inhibit CO2 formation thus acting as an inhibitor.
50.
Industrial catalysts
Broadly groupedinto three categories:
1.Bulk catalysts : When the entire catalyst consists of the
catalytically active substance ,then the solid catalyst is
called a bulk catalyst. Examples include silica alumina
catalysts for catalytic cracking; iron- molybdate for
oxidation of methanol to formaldehyde; iron doped with
alumina and potassium oxide for the synthesis of
ammonia.
2. Supported catalysts: In supported catalysts, the
catalytically active materials are dispersed over the high
surface area support material. For example,
hydrodesulphurization is carried out over molybdenum
oxide supported on alumina.
51.
Industrial catalysts
3. Mixedagglomerates :
• These catalysts are agglomerated mixture of active
substance and support. These type of catalysts are
used less frequently.
52.
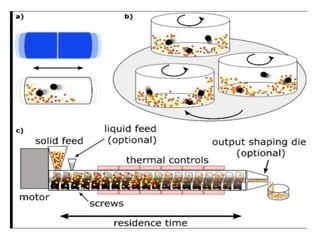
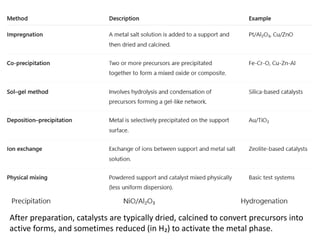
Preparation of solidcatalyst
The catalyst preparation methods can broadly categorized
as follows :
1. Bulk preparation process: Bulk catalysts and supports
are prepared by this method. Bulk preparation is mainly
done by the following methods :
a. Precipitation process
b. Sol gel process
53.
Preparation of solidcatalyst
2. Impregnation process: Supports are first prepared by
bulk preparation methods and then impregnated with the
catalytically active material.
The active materials can be deposited on the supports by
various methods. Most of the methods involve aqueous
solutions and liquid solid interface.
In some cases, deposition is done from the gas phase and
involves gas- solid interface.
54.
3. Physical mixing:
Mixed agglomerated catalysts are prepared by this
method.
These catalysts are prepared by physically mixing the active
substances with a powdered support or precursors of
support in ball mill.
The final mixture is then agglomerated and activated.
56.
3. Physical mixing:
Mixed agglomerated catalysts are solid catalyst systems in
which two or more catalytic components or phases are
physically combined and agglomerated (i.e., clustered or
compacted) into a single solid structure—typically as
pellets, granules, tablets, or extrudates.
Unlike simple physical mixtures, these systems are
designed so that the components interact closely, either
synergistically or spatially, to improve overall catalytic
performance.
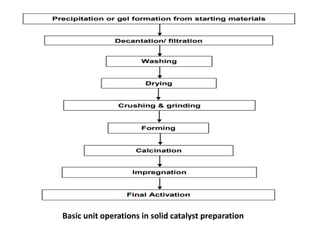
Precipitation or GelFormation from Starting Materials
•What happens: Metal salts (e.g., nitrates, sulfates) are reacted
with a precipitating agent (e.g., NaOH, NH₄OH, carbonates) to
form insoluble hydroxides, carbonates, or oxides, or a sol–gel
network.
•Purpose: Converts soluble precursors into solid particles that can
become the active component or support.
•Example: Co-precipitation of Cu²⁺, Zn²⁺, and Al³⁺ to form a Cu-Zn-
Al mixed hydroxide.
Decantation / Filtration
•What happens: The solid precipitate or gel is separated from the
liquid (mother liquor) by gravity (decantation) or filtration.
•Purpose: To isolate the solid and remove soluble by-products,
unreacted salts, or excess reagents.
59.
Washing
•What happens: Thesolid is washed multiple times with distilled
or deionized water.
•Purpose: Removes impurities, residual ions (e.g., Na⁺, Cl⁻, NO₃⁻),
and undesirable side-products that could interfere with catalytic
activity or poison the catalyst.
Drying
•What happens: Moisture is removed from the washed solid by
heating at low temperatures (e.g., 100–120 °C).
•Purpose: Prepares the solid for further processing and
prevents agglomeration or reaction during calcination.
•Typical equipment: Oven, vacuum dryer, or rotary dryer.
60.
Crushing & Grinding
•Whathappens: The dried solid is mechanically broken down into
fine powder using mortar, mill, or grinders.
•Purpose: Ensures uniform particle size and better dispersion in
subsequent steps like impregnation.
Impregnation (optional, if metal active phase is added later)
•What happens: The support is soaked or treated with a solution
of the active metal precursor (e.g., H₂PtCl₆, Ni(NO₃)₂).
•Purpose: Deposits metal ions or complexes on the surface or
within the pores of the support.
•Methods:
• Incipient wetness: Just enough liquid to fill the pores
• Wet impregnation: Excess solution
61.
Calcination
•What happens: Theimpregnated or precipitated material is heated
in air or oxygen at high temperatures (300–800 °C).
•Purpose:
• Converts precursors to oxides
• Removes organic matter, nitrates, or other volatiles
• Improves mechanical strength and porosity
• Anchors active metal particles on the support
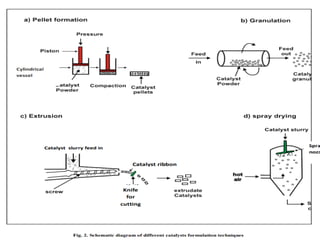
Forming
•What happens: The powder is shaped into pellets, extrudates,
tablets, spheres, or monoliths.
•Purpose: Makes the catalyst suitable for use in industrial reactors
by:
• Increasing mechanical strength
• Reducing pressure drop
• Ensuring good flow and heat transfer
•Methods: Pelletizing, extrusion, tableting, spray drying
62.
Final Activation
•What happens:The catalyst is subjected to reduction (in
H₂), sulfidation, or other activation depending on the
system.
•Purpose: Converts the metal oxide to active metal, e.g.,
NiO → Ni⁰, or forms the desired active phase.
•Typical conditions: H₂ gas at elevated temperatures (e.g.,
300–500 °C)
63.
After preparation, catalystsare typically dried, calcined to convert precursors into
active forms, and sometimes reduced (in H₂) to activate the metal phase.
64.
Precipitation and coprecipitation
•Widely used methods for the preparation of heterogeneous
catalysts, especially oxide- and mixed-oxide-based systems such
as Cu/ZnO/Al₂O₃, Ni/Al₂O₃, Co–Fe oxides catalysts.
• They allow good control over surface area, particle size, and
composition—all of which influence catalytic activity.
65.
Precipitation Method
A processin which a soluble precursor salt of the desired
catalytic component is converted into an insoluble
compound (often a hydroxide, carbonate, or basic salt)
by adding a precipitating agent, usually under controlled
pH and temperature.
The precipitate is later filtered, washed, dried, calcined,
and optionally reduced to obtain the final catalyst.
66.
Steps:
1.Preparation of precursorsolution
Dissolve a metal salt (e.g., Ni(NO₃)₂, Cu(NO₃)₂) in water.
2.Addition of precipitating agent
Common agents: Na₂CO₃, NaOH, (NH₄)₂CO₃, urea (thermal hydrolysis).
3.pH control
Maintain constant pH to control precipitation rate and particle size.
4.Aging
Allow precipitate to mature so particles grow and crystallinity improves.
5.Separation
Filter or decant the solid phase.
6.Washing
Remove residual ions (Na⁺, NO₃⁻, Cl⁻) that could poison the catalyst.
7.Drying
Usually at 100–120 °C to remove water.
8.Calcination
300–800 °C to decompose hydroxides/carbonates to oxides.
9.Activation
Reduction in H₂ if metallic state is required.
67.
2. Coprecipitation Method
Similarto precipitation, but two or more metal precursors are
precipitated simultaneously from a homogeneous solution by the
addition of a precipitating agent. This ensures intimate mixing of
different metals at the atomic or nanoscale level.
Advantages over precipitation:
•Produces uniform distribution of metals in the support or mixed
oxide.
•Better metal–support interaction.
•Enhanced thermal stability and resistance to sintering.
•Often higher surface area and dispersion.
68.
Summary:
•Precipitation:
Single metal precursor→ insoluble salt → oxide/metal catalyst.
•Coprecipitation:
•Multiple precursors precipitated together → intimate mixing →
better dispersion and synergy.
•Both methods are simple, scalable, and widely used for industrial
catalyst preparation.
69.
1. SiO2-Al2O3
SiO2-Al2O3 isused in catalytic cracking process and is also used
as support for active metals in various applications. Preparation
of dual oxides by coprecipitation is similar to precipitation of
single oxide. At pH 6 (at 50 0C) the precipitation of both silica
and alumina sols begins, and gelation takes places.
Coprecipitation Method examples
2. NiO-Al2O3
NiO-Al2O3 is used for hydrogenation and methanation reactions.
Although this catalyst can be produced by other route,
coprecipitation method of preparation is also done to increase the
intimate interaction between active metal and support.
70.
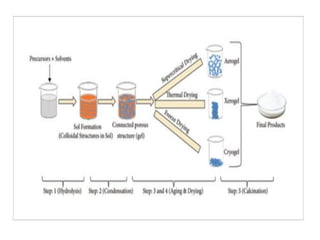
Sol gel method
•Is a chemical technique used to create materials, often
nanoparticles or thin films, by forming a gel from a solution (sol)
• widely used technique in catalysis for preparing high-surface-area
catalyst supports and active materials with well-controlled
properties.
• It allows the synthesis of homogeneous, porous, and thermally
stable materials such as silica, alumina, titania, and mixed oxides,
which are often used as supports or even as active catalysts
themselves.
71.
In the solgel process,
• Initially a stable colloidal solution called sol is formed.
• The sol is a liquid suspension of solid particles ranging in
size from 1 nm to 1 micron.
• It can be obtained by hydrolysis and partial
condensation of precursors such as an inorganic salt or
a metal alkoxide. The further condensation of sol
particles into a three dimensional network produces a
gel material.
• The gel is a diphasic material in which the solids
encapsulate the solvent.
72.
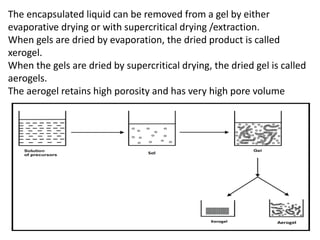
The encapsulated liquidcan be removed from a gel by either
evaporative drying or with supercritical drying /extraction.
When gels are dried by evaporation, the dried product is called
xerogel.
When the gels are dried by supercritical drying, the dried gel is called
aerogels.
The aerogel retains high porosity and has very high pore volume
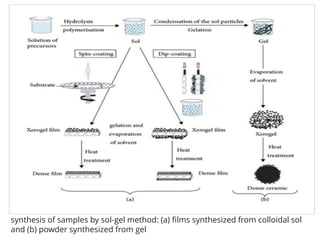
74.
synthesis of samplesby sol-gel method: (a) films synthesized from colloidal sol
and (b) powder synthesized from gel
75.
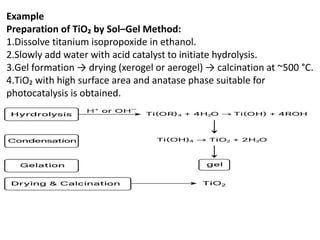
Example
Preparation of TiO₂by Sol–Gel Method:
1.Dissolve titanium isopropoxide in ethanol.
2.Slowly add water with acid catalyst to initiate hydrolysis.
3.Gel formation → drying (xerogel or aerogel) → calcination at ~500 °C.
4.TiO₂ with high surface area and anatase phase suitable for
photocatalysis is obtained.
77.
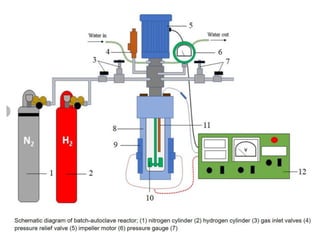
Catalytic Hydrogenation
Catalytic hydrogenationis a chemical reaction in which
hydrogen (H₂) is added to unsaturated organic
compounds in the presence of a catalyst.
The process is widely used in:
•Petrochemicals (fuel upgrading)
•Fine chemicals (flavors, fragrances, dyes)
•Pharmaceuticals (drug synthesis)
•Food industry (vegetable oil hydrogenation)
78.
Types of CatalyticHydrogenation
A. Heterogeneous Catalytic Hydrogenation
•Catalyst phase: Solid
•Reactants phase: Gas or liquid
•Common Catalysts:
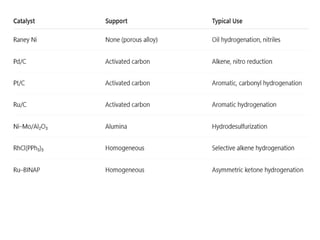
• Ni (Raney Ni, supported Ni)
• Pd/C (palladium on carbon)
• Pt/C (platinum on carbon)
• Ru/C, Rh/C
•Examples:
• Alkene → Alkane
• Nitrobenzene → Aniline
• Benzene → Cyclohexane
79.
B. Homogeneous CatalyticHydrogenation
•Catalyst phase: Same phase as reactants (solution)
•Common Catalysts:
• Wilkinson’s catalyst [RhCl(PPh₃)₃]
• Chiral Rh/Ru complexes (for asymmetric hydrogenation)
•Examples:
• Selective alkene hydrogenation in complex molecules
• Asymmetric hydrogenation for chiral drug synthesis (e.g., L-
DOPA)
80.
Catalyst Selection Factors
•Activity– How fast the reaction proceeds.
•Selectivity – Preferential hydrogenation of specific bonds
(C=C vs C=O).
•Stability – Resistance to deactivation (poisoning,
sintering).
•Cost & Availability – Ni is cheaper, Pt/Pd are expensive.
•Reusability & Regeneration – Ease of catalyst recovery.
84.
Recent Developments
•Nanostructured catalysts– higher activity and selectivity.
•Bimetallic catalysts – synergistic effects (e.g., Pd–Ag, Pt–Sn).
•Green hydrogenation – using renewable H₂ from electrolysis.
•Metal-free hydrogenation – using frustrated Lewis pairs (FLPs).
Frustrated Lewis pairs (FLPs)
Combinations of a Lewis acid and a Lewis base
due to steric hindrance or electronic effects, cannot form a stable
adduct.
This “frustration” leaves both species reactive enough to
cooperatively activate small molecules such as H₂ — without the
need for transition metals.
The Lewis base abstracts H⁺.
The Lewis acid accepts H⁻.
•Hydrogenation of Oils
•Catalyst: Ni or Pd.
• Produces margarine and specialty fats.
•Asymmetric Hydrogenation
• Catalyst: Rh or Ru complexes with chiral ligands.
• Produces enantiopure pharmaceuticals.
Environmental Applications
1.Hydrogenation of CO/CO₂
1. CO₂ + 3H₂ → CH₃OH (methanol).
2. CO + H₂O → CO₂ + H₂ (Water–Gas Shift Reaction).
3. Catalysts: Cu/ZnO, Ni.
88.
Oxidative Dehydrogenation
Oxidative dehydrogenation(ODH) is a catalytic process where
hydrogen atoms are removed from hydrocarbons in the presence
of oxygen, producing olefins or aromatic intermediates.
It is an alternative to conventional steam cracking or catalytic
dehydrogenation, often offering lower temperatures and reduced
coke formation.
Aromatic Hydrocarbon Upgrading
•ODHof Alkyl Aromatics
• Example: p-Xylene → p-Toluic acid intermediates for
terephthalic acid production.
• Used in specialty chemical synthesis.
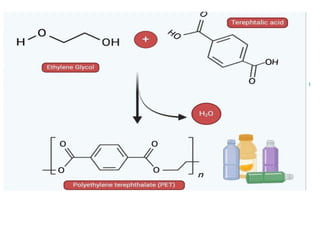
Terephthalic acid (C₆H₄(COOH)₂) is the primary raw
material for PET used in:
•Textile fibers
•PET bottles
•Packaging films
Advantages of ODHover Conventional Dehydrogenation
•Lower reaction temperature (350–500 °C vs. 600–900 °C).
•Avoids equilibrium limitations of non-oxidative dehydrogenation.
•Reduced coke deposition → longer catalyst life.
•Potential for energy savings and lower CO₂ emissions.
Industrial Relevance
•Ethylene – world’s largest volume organic chemical (>200 million
tonnes/year).
•Propylene – demand for polypropylene and propylene derivatives
growing rapidly.
•Butadiene – essential for tire and rubber industry.
•Styrene – important for plastics, resins, and insulation materials.
95.
Oxidative Organic Transformations
Theseare oxygen-assisted catalytic reactions used to make valuable
fine chemicals and pharmaceutical intermediates.
Examples:
Industrial Significance ofODH
•Petrochemical industry – production of ethylene, propylene,
butadiene via ODH.
•Polymer industry – monomers like styrene, maleic anhydride,
acrylic acid.
•Fine chemical industry – oxidation to aldehydes, ketones, and
acids for fragrances, solvents, and pharmaceuticals.
•Energy sector – partial oxidation for syngas production.
•Environmental catalysis – VOC oxidation, CO oxidation in
automotive converters.
98.
The oxidative alkylationof amines is an important organic
transformation in which an amine is alkylated via oxidative
activation of a substrate, often under catalytic conditions.
It’s frequently applied for the synthesis of secondary and
tertiary amines and in C–N bond formation in
pharmaceuticals, agrochemicals, and fine chemicals.
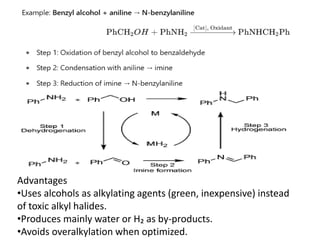
Oxidative alkylation of amines
99.
General Concept
Oxidative alkylationcouples an amine (R¹NH₂ or R¹R²NH) with an
alkylating agent (often an alcohol or alkyl halide precursor) in the
presence of an oxidant or via dehydrogenative activation.
Typical pathways:
1.Alcohol Oxidation Pathway:
1. Alcohol → aldehyde (via oxidation)
2. Aldehyde + amine → imine (condensation)
3. Imine → alkylated amine (hydrogenation or hydride transfer)
This is known as the Borrowing Hydrogen (BH) methodology or
Hydrogen Autotransfer.
100.
Advantages
•Uses alcohols asalkylating agents (green, inexpensive) instead
of toxic alkyl halides.
•Produces mainly water or H₂ as by-products.
•Avoids overalkylation when optimized.
101.
2. Direct OxidativeC–H Activation:
Direct oxidative C–H activation is a catalytic process that
transforms inert C–H bonds into C–X bonds (X = C, N, O, halogen)
without pre-functionalization of the substrate.
Instead of requiring pre-activated substrates (like halides or
organometallics), the reaction directly uses C–H bonds as
functional handles.
103.
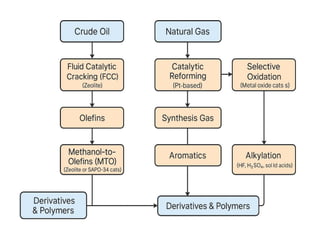
Catalysis in theProduction of petrochemicals
Catalysis plays a central role in the production of petrochemicals
because most conversion processes require catalysts to achieve
high selectivity, lower energy consumption, and economic viability.
Role of Catalysis
• Lower activation energy → allows reactions at lower
temperatures/pressures.
• Improve selectivity → maximize desired product yield, minimize
by-products.
• Enable specific transformations → cracking, reforming,
isomerization, alkylation, oxidation, hydrogenation.
Important Catalytic Processesin Petrochemicals
Feedstocks
•Crude Oil → main source of heavier hydrocarbons
•Natural Gas → mainly methane, ethane, propane, butane
From Crude Oil
• Fluid Catalytic Cracking (FCC) – uses zeolite catalysts to break large
hydrocarbon molecules into lighter olefins and gasoline-range
products.
• Catalytic Reforming – uses Pt-based catalysts to convert naphtha
into aromatics (benzene, toluene, xylenes) and hydrogen.
• Hydroprocessing – removes sulfur, nitrogen, and other impurities
before further catalytic steps.
107.
From Natural Gas
•SteamReforming – uses Ni-based catalysts to convert methane +
steam into synthesis gas (syngas: H₂ + CO), which is the basis for
methanol, ammonia, and other intermediates.
•Selective Oxidation – uses metal oxide catalysts to produce partial
oxidation products (e.g., ethylene oxide from ethylene).
Product Streams
•Olefins → polyethylene, polypropylene, acrylonitrile, ethylene
oxide.
•Aromatics → styrene, polyester, nylon intermediates.
•Synthesis Gas → methanol, ammonia, synthetic fuels.
•Derivatives & Polymers → plastics, fibers, rubbers, resins.
108.
Overview
•Syngas = CO+ H₂ (produced from coal gasification, natural gas
reforming, or biomass gasification)
• Convert syngas into aromatic hydrocarbons (benzene, toluene,
xylenes ) for petrochemical feedstocks.
• Requires selective C–C bond formation and ring aromatization
from C₁ building blocks.
Two-Step Route
1.Fischer–Tropsch Synthesis (FTS)
1. Syngas → light olefins (C₂–C₄) or paraffins.
2. Catalyst: Fe- or Co-based.
3. Reaction:
Syngas
is a mixtureof carbon monoxide (CO) and hydrogen (H₂), sometimes
with carbon dioxide (CO₂), methane (CH₄), or nitrogen (N₂) depending
on the production method. It is not a fuel itself but a precursor to
many fuels and chemicals.
111.
Uses of Syngas
•Fischer–TropschProcess: → Liquid hydrocarbons (diesel, kerosene)
•Methanol Production: CO + 2H₂ → CH₃OH
•Ammonia Production: N₂ + 3H₂ → 2NH₃ (via hydrogen from syngas)
•Hydrogen Production: Purified from syngas
•Dimethyl ether (DME): Alternative clean fuel
112.
Ammonia Synthesis –Haber–Bosch Process
• H₂ and N₂ are compressed to 150–300 atm and heated to 400–
500°C.
• Iron-based catalyst (Fe with promoters like K₂O, Al₂O₃, CaO) is used.
• The reaction occurs in a loop reactor; unreacted gases are recycled.
A loop reactor is an essential component of modern ammonia
plants, designed to handle the high-pressure, high-temperature,
and reversible
114.
Mobil Process: Conversionof Methanol to Gasoline (MTG
Process)
Mobil Methanol-to-Gasoline (MTG) process is a catalytic route
developed by Mobil Oil Corporation to produce high-octane
gasoline from methanol.
115.
Key Steps ofthe MTG Process
1. Methanol Dehydration
•Reaction:
2. DME and Methanol Conversion to Olefins
•Catalyst: ZSM-5 (zeolite)
•Produces light olefins like ethylene, propylene, and butenes through cracking
and rearrangement.
3. Oligomerization and Cyclization
•Olefins are oligomerized (combined into longer chains), cyclized to form cyclic
hydrocarbons (naphthenes), and aromatized.
4. Hydrogen Transfer & Aromatization
•Hydrogen transfer leads to saturation and aromatic formation.
•Major products: alkanes, isoparaffins, aromatics — similar to gasoline.
5. Product Separation
•Final products are cooled and separated into gasoline, light gases, and water.
116.
Catalysis for Polymerization
Useof catalysts to initiate and control the formation of
polymers from monomers.
Catalysts enable faster, more selective, and often greener
polymerization processes, which are essential in producing
plastics, rubbers, and advanced materials.
Functions of Catalysts in Polymerization
•Control polymer chain length (molecular weight).
•Influence tacticity (stereochemistry of polymer chains).
•Improve selectivity for specific monomers.
•Enable controlled polymerization.
117.
Types of Polymerizationand Catalysts
1. Addition (Chain-Growth) Polymerization
•Monomers: Unsaturated compounds like ethene, styrene,
acrylonitrile.
•Catalysts:
• Ziegler–Natta catalysts → For stereoregular polymers like
polypropylene.
• Metallocenes → High control over tacticity
(isotactic/syndiotactic polymers).
• Free Radical Initiators → Benzoyl peroxide,
• Anionic or Cationic Catalysts → For polystyrene,
polyisobutylene.
Anionic Catalysts inPolymerization
Initiation involves a nucleophilic species (anion or Lewis base)
attacking an electron-deficient monomer (often containing an
electron-withdrawing group).
•Initiator examples:
• Alkali metal alkoxides (RO⁻ M⁺)
• Organolithium reagents (R–Li)
• Sodium naphthalide
• Anionic PTC catalysts (e.g., PEG-Na⁺ complexes)
•Suitable monomers:
• Styrene
• Acrylonitrile
• Methacrylates
• Butadiene, Isoprene
Example reaction:
Styrene + n-BuLi → Polystyrene
121.
Cationic Catalysts inPolymerization
Initiation involves an electrophilic species (cation or Lewis acid)
attacking an electron-rich monomer (usually alkenes with electron-
donating groups).
•Catalyst examples:
• BF₃·OEt₂ (boron trifluoride etherate)
• AlCl₃, TiCl₄
• H₂SO₄, HClO₄
• Metal halides with co-catalysts
•Suitable monomers:
• Isobutene
• Vinyl ethers
• Styrene (with activating substituents)
Example reaction:
Isobutene + BF₃ → Polyisobutene
122.
Condensation (Step-Growth) Polymerization
•Monomers:Diacids + Diols, Diamines + Diacids, etc.
•Catalysts:
• Acid Catalysts (H₂SO₄, p-TsOH) → For polyesters like PET.
• Base Catalysts (NaOH) → For polyamides and some
polyurethanes.
• Metal Complexes → Used for ring-opening polymerizations
(e.g., lactide to PLA).
Ring-Opening Polymerization (ROP)
•Used to make biodegradable polymers like polylactide (PLA) and
polycaprolactone.
•Catalysts:
• Tin(II) octoate (Sn(Oct)₂)
• Aluminum and lanthanide complexes
• Organocatalysts (e.g., DBU, TBD)
123.
Green Catalysis inPolymerization
•Metal-free organocatalysts → Biodegradable and non-toxic.
•Enzymatic polymerization → Mild conditions, sustainable.
•CO₂-based polymerization → Using CO₂ to form polycarbonates.
124.
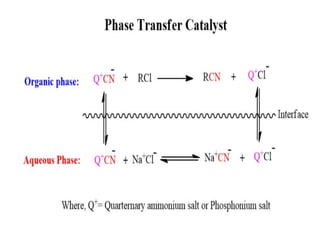
Phase Transfer Catalysis(PTC)
Is a technique in chemistry that allows a reaction to occur
between reactants present in different immiscible phases
(usually organic and aqueous) by using a phase transfer
catalyst—a substance that can transport reactants between the
two phases.
125.
Why PTC isNeeded
• Many organic reactions involve an ionic reactant (e.g., anions like
OH⁻, CN⁻, or halides) that is soluble in the aqueous phase but not in
the organic phase where the other reactant resides.
• Without a transfer mechanism, these reactions would be slow
because the reactants are separated by the phase boundary.
• Phase transfer catalysts carry the ionic species into the organic
phase where it can react with the organic substrate.
126.
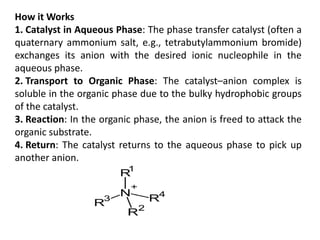
How it Works
1.Catalyst in Aqueous Phase: The phase transfer catalyst (often a
quaternary ammonium salt, e.g., tetrabutylammonium bromide)
exchanges its anion with the desired ionic nucleophile in the
aqueous phase.
2. Transport to Organic Phase: The catalyst–anion complex is
soluble in the organic phase due to the bulky hydrophobic groups
of the catalyst.
3. Reaction: In the organic phase, the anion is freed to attack the
organic substrate.
4. Return: The catalyst returns to the aqueous phase to pick up
another anion.
128.
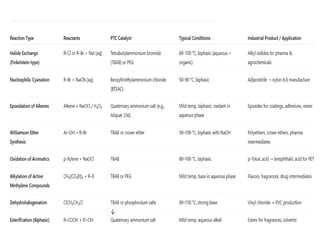
Common Catalysts
• Quaternaryammonium salts: e.g., tetra-n-butylammonium
bromide (TBAB), benzyltriethylammonium chloride (BTEAC)
• Phosphonium salts: e.g., tetrabutylphosphonium bromide
• Crown ethers: e.g., 18-crown-6 (complex alkali metal cations)
• Polyethylene glycols (PEGs): act via solubilizing metal cations
129.
Applications
•Nucleophilic substitution: e.g.,alkyl halide + CN⁻ → nitrile
•Oxidations: e.g., aqueous NaOCl with organic substrates
•Esterification in biphasic systems
•Environmental: detoxification of waste streams
130.
Advantages
•Faster reactions atmild temperatures
•Higher yields in biphasic systems
•Avoids expensive or hazardous solvents
•Allows reactions with inexpensive aqueous reagents
Limitations
•Catalyst contamination in product
•Limited to systems where ionic transfer is possible
•Not always effective for bulky or highly hydrophilic ions
132.
Biocatalysis
is the useof natural catalysts—primarily enzymes, but also
whole cells—to carry out chemical reactions, often under
mild and environmentally friendly conditions.
Advantages
1.Mild conditions — often near room temperature, neutral pH,
atmospheric pressure.
2.High selectivity — reduces side products, often eliminates need for
protecting groups.
3.Sustainability — biodegradable catalysts, less hazardous waste.
4.Renewable source — enzymes derived from plants, animals, or
microbes.
133.
Limitations
•Narrow temperature orpH range for enzyme stability.
•Sensitivity to solvents or inhibitors.
•Sometimes slower than chemical catalysts.
Types of Biocatalysts
1.Enzymes
1. Hydrolases (lipases, proteases, esterases)
2. Oxidoreductases (alcohol dehydrogenase, peroxidases)
3. Lyases, isomerases, transferases, ligases
2.Whole cells
1. Microbial cells with multiple enzyme pathways (E. coli, yeast,
fungi)
134.
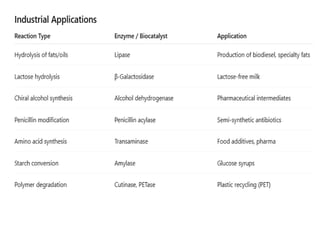
Important industrial-scale biocatalyticreactions
Biodiesel Production
•Reaction: Transesterification of triglycerides with methanol
•Biocatalyst: Lipase (e.g., Candida antarctica lipase B)
•Conditions: Mild temp (30–50 °C), solvent-free or in organic solvent
•Application: Biodiesel from vegetable oils and animal fats.






































































![B. Homogeneous Catalytic Hydrogenation
•Catalyst phase: Same phase as reactants (solution)
•Common Catalysts:
• Wilkinson’s catalyst [RhCl(PPh₃)₃]
• Chiral Rh/Ru complexes (for asymmetric hydrogenation)
•Examples:
• Selective alkene hydrogenation in complex molecules
• Asymmetric hydrogenation for chiral drug synthesis (e.g., L-
DOPA)](https://image.slidesharecdn.com/catalysis-250908113159-f510b027/85/Catalysis-Introduction-Mechanistic-insights-Applications-79-320.jpg)