This document discusses chemical kinetics and reaction rates. It begins with an introduction to chemical kinetics and defines reaction rate. It then discusses factors that affect reaction rates such as nature of reactants, concentration, temperature, and catalysts. It describes different types of reaction rates and how they are measured. The document also covers rate laws, determining rate orders experimentally, and integrated and differential rate equations for zero, first, and second order reactions. It concludes with an overview of rate theories including the Arrhenius equation.







![Types of reaction rate
• Instantaneous rate – rate at a specific time
• Average rate – ∆[A] over a specific time interval
• Initial rate – instantaneous rate at t = 0
• Note: Rates and rate laws are not based on
stoichiometry!! They must be determined
experimentally.
8](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-8-320.jpg)





![• Rate has units of moles per liter per unit time
- M s-1, M h-1
• Consider the hypothetical reaction
aA + bB cC + dD
• We can write
Reaction rates and stoichiometry
14
t
D
dt
C
c
t
B
bt
A
a
r
][1][1
][1][1](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-14-320.jpg)


![Rate law
• Consider the following reaction
aA + bB products
• Rate Law: equation describing the
relationship between the reaction rate and
concentration of a reactant or reactants
Rate = k[A]m[B]n
where k is called the rate constant
17](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-17-320.jpg)
![Concentration and rate
aA + bB → cC + dD
• General form of rate law:
rate = k[A]m[B]n
[A], [B] – concentration, in M or P
k – rate constant; units vary
m, n – reaction orders
• Reaction orders and, thus, rate laws must be
determined EXPERIMENTALLY!!!
– Note: m ≠ a and n ≠ b
– Overall order = sum of individual orders
18](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-18-320.jpg)
![Reaction order
• Rate = k[A][B]0 m = 1 and n = 0
- reaction is first order in A and zero order in B
- overall order = 1 + 0 = 1
- usually written: Rate = k[A]
• The values of the reaction order must be determined
experimentally; they cannot be found by looking at
the equation, i.e., the stoichiometry of the reaction
19](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-19-320.jpg)


![Reaction orders
For the reaction: A →B, the rate law is:
rate = k[A]m
Order (m) ∆[A] by a factor of Rate increases by
Zero (0) 2, 4, 15, ½, etc. None
1st (1)
2 2X
3 3X
2nd (2)
2 4X
3 9X
½ ¼X
22](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-22-320.jpg)
![Example
1. What is the order with respect to NO?
2. What is the order with respect to H2?
3. What is the overall order?
4. If [NO] is doubled, what is the effect on the reaction
rate?
5. If [H2] is halved, what is the effect on the reaction
rate?
6. What are the units of k?
23
2
1
3
quadrupled
halved
M-2-s-2
2 NO(g) + 2 H2(g) → N2(g) + 2 H2O(g)
rate = k[NO]2[H2]](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-23-320.jpg)
![24
Rate = 9x10-3 (h-1) x 0.02 (M) = 1.8x10-5 (M-h-1)
d[Cl]/dt = rate = 1.8x10-5 (M-h-1)
What is the rate of Cl- production under
these conditions?
Calculate the rate of reaction when the
concentration of PtCl2(NH3)2 is 2.0x10-2 M.
PtCl2(NH3)2 + H2O PtCl(H2O)(NH3)2 + Cl-
Example](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-24-320.jpg)
![Calculate rate when [NO] = 0.025 M and [H2] = 0.015 M.
25
Rate = 6x10-4 (M-2-s-1)x0.025 M x 0.015 M
=225 (M-s-1)
2 NO(g) + 2 H2(g) → N2(g) + 2 H2O(g)
rate = k[NO]2[H2] k = 6.0 x 104 M-2s-1 @1000K
Example](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-25-320.jpg)

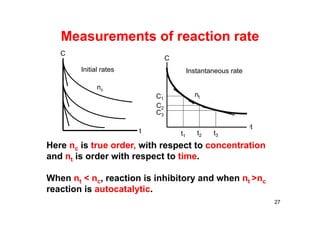
![In this reaction, the
concentration of butyl
chloride, C4H9Cl, is
measured at various
times.
C4H9Cl(aq) + H2O(l) → C4H9OH(aq) + HCl(aq)
28
Time, s [C4H9Cl], M
0 0.1000
50 0.0905
100 0.0820
150 0.0741
200 0.0671
300 0.0549
400 0.0448
500 0.0368
800 0.0200
10,000 0](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-28-320.jpg)
![29
Time, s [C4H9Cl], M Average rate, M/s
0 0.1000
50 0.0905 1.9E-4
100 0.0820 1.7E-4
150 0.0741 1.6E-4
200 0.0671 1.4E-4
300 0.0549 1.22E-4
400 0.0448 1.01E-4
500 0.0368 0.8E-4
800 0.0200 0.56E-4
10,000 0](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-29-320.jpg)








![First order
]A)[I,T(k
dt
]A[d
ν
R
A
1
t)I,T(k
o e]A[]A[
t
.
)I,T(k
]Alog[]Alog[ o
3032
1.0
0.1
0.01
0.001
1 2 3
t
Log [A]
40](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-40-320.jpg)
![Second order
2
A
]A)[T(k
dt
]A[d1
R
t)T(kν
]A[]A[
i
0
11
41
t)T(k]A[]B[
]A[
]B[
ln
]A[
]B[]A[
ln oAoB
o
oAoAoB
](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-41-320.jpg)
![Third order
]C][B][A)[T(k
dt
]A[d1
R
A
1CBA
ooo ]C[]B[]A[
t)T(k2
]A[
1
]A[
1
2
o
2
42](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-42-320.jpg)
![Summary Rate Equations
43
Rate = k[A]n; [A] = concentration at time t, [Ao] = initial concentration, [X] =
product conc.
[A0]-[A] = kt, [X] = kt
ln[A0] - ln[A] = kt, ln[A0] - ln([Ao] - [X]) = kt
0 order k = M/s
1st order k = 1/s
2nd order
k = M-1-s-1
3rd order k = M-2-s-1](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-43-320.jpg)

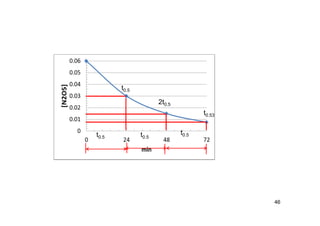
![Half-Life
45
• Half-life
– A =Aoe-t
– = ln2/t1/2
– If a rate half life is known,
fraction reacted or
remaining can be
calculated
(CH3)2N2(g) N2(g) + C2H6(g)
Time Pressure (torr)
0 36.2
30 46.5
Ct =Coe-kt
kt0.5=ln(2)
to.5 = - [ln(2) t]/[ln(Ct/Co)]
to.5 = - [ln(2) (30)]/[ln(25.9/36.5)]=62.1 (min)
t A B C
0 36.2 0 0
3036.2(1-x) 36.2x 36.2x
46.5 = 36.2(1-x+2x)=36.2(1+x)=36.2+36.2x
46.5-36.2 = 36.2x x = 0.285
A = 36.5(1-0.285) = 25.9](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-45-320.jpg)


![ARRHENIUS’ EQUATION
49
E1
E2
E=Ea
Initial state
Final state
Activated state; X*
E1
E2
E=Ea
Initial state
Final state
Activated state; X*
DCBA kk
11
]][[]][[ 11 DCkBAk
]][[
]][[
1
1
BA
DC
k
k
Kc
11 EEE
2
11 lnln
RT
E
dt
kd
dt
kd
1
ln
2
11
RT
E
dt
kd
1
ln
2
11
RT
E
dt
kd
2
ln
RT
E
dT
kd a
C
RT
E
k a
ln C = constant](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-49-320.jpg)


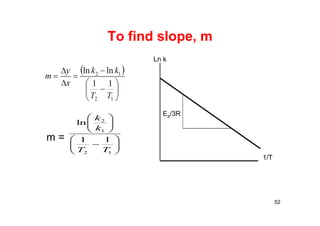
![Arrhenius’ Equation
• This is Arrhenius’ Equation
• Can be arranged in the form of a straight line
• ln k = (-Ea/R)(1/T) + ln A
• Plot ln k vs. 1/T slope = -Ea/R
1.0
0.1
0.01
0.001
1 2 3
1/T
Log [k]
EA/2.303
= dlog K/d(1/T)
Diffusion
regime
Reaction
regime
Ea < 5 kcal/mol: diffusion
control reaction
Ea > 5 kcal/mol; reactive
control
53](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-53-320.jpg)



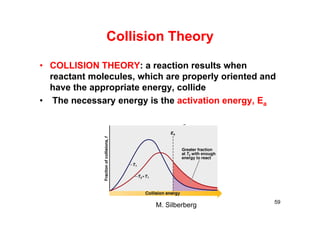

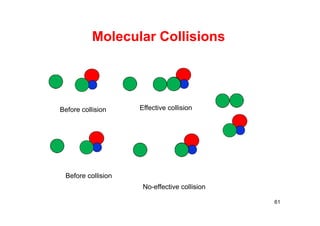
![Collision Theory
RT
E
AAezv
22
AA ncd2
2
1
z
m
T
dn2
m
T8
nd2
2
1
z 2222
AA
RT
E
BA
BA2
ABBA e
mm
mm
T8dnnv
2
1
62
molecules/(cm3-s)
mπ
Tκ
c
8
= average velocity of each molecule
m: mass of each molecules
k = Boltzmann constant
dAB: average distance, sum of radii
two identical gas molecules colliding with each other at a velocity,
v (molecules/cm3-s) [N.C. Leuis (1918) and Eyring (1935)]
zAA = # collision/(s-cm3)](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-62-320.jpg)




![Transition State Theory
]][[
*][**
*
BA
AB
aa
a
K
BA
AB
BA
AB
]][[*
*
BAKr
BA
AB
RT
G
eK
*
*
T
= 6.624x10-27 erg/s
70
K = 1.38x10-16 erg/oK
A + B = AB* C
r = [AB*]; (s-1); [AB*](molecules/cm3)](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-70-320.jpg)






![Elementary Steps and Molecularity
Molecularity is the number of molecules reacting.
77
Molecularity Elementary Reaction Rate Law
Unimolecular A products Rate = k[A]
Bimolecular A + A products Rate = k[A]2
Bimolecular A + B products Rate = k[A][B]
Trimolecular A + A + A products Rate = k[A]3
Trimolecular A + A + products Rate = k[A]2[B]
Trimolecular A + B + C products Rate = k[A][B][C]](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-77-320.jpg)

![79
2N2O5 4NO + O2
N2O5 NO2 + NO3
k1
k-1
NO3 + NO NO + NO2 + O2
k2
NO3 + NO 2NO
k3
0]][[]][[
][
3332 NONOkNONOk
dt
NOd
][
]][[
][
33
232
NOk
NONOk
NO
0]][[]][[]][[][
][
32133232521
3
NONOkNONOkNONOkONk
dt
NOd
]][[]][[]][[
][
][
32133232
521
3
NONOkNONOkNONOk
ONk
NO
](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-79-320.jpg)
![80
])[2(
][
][])[(
][
][][][
][
][
212
521
2
3
2
3212
521
21322
521
3
NOkk
ONk
NO
k
k
kNOkk
ONk
NOkNOkNOk
ONk
NO
]['
)2(
][
])[2(
]][[
]][[
][
52
12
52321
212
252321
232
2
ONk
kk
ONkkk
NOkk
NOONkkk
NONOk
dt
Od
r
](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-80-320.jpg)


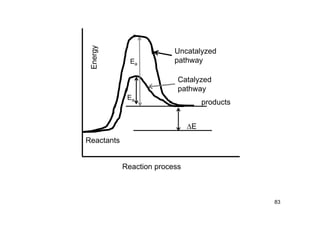


![86
Catalytic efficiency,
= kcat/KM
= k2/[(k-1+k2)/k1]
max, = k1; if k2 >>k-1
k1 = rate of formation of ES
Diffusion limit ~ 108 – 109 M-1-s-1
For enzyme sized molecules at room
temperature
Decomposition of hydrogen peroxide
= 4x108 M-1-s-1
Turnover number or catalytic constant
= number of catalytic cycles performed by the activate
Site in a given time intervals divided by the duration of that](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-86-320.jpg)
![87
Lineweaver Burk Plot ][
][max
SK
Sv
v
M
SSv
K
v
M 111
max
maxmax v
S
v
K
v
S M
MM K
v
K
v
S
v
max
1/vmax
KM/vmax
1/v
1/[S]
vmax/KM
1/KM
v/[S]
v
KM/vmax
1/vmax
[S]/v
[S]](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-87-320.jpg)

![90
Competitive Inhibition
0][][]][[
][
211 ESkESkSEk
dt
ESd
0][][]][[
][
433 EIkEIkIEk
dt
EId
MK
k
kk
ES
SE
1
21
IK
EI
IE
][][][ EIESEEo
IM
o
K
IE
K
SE
EE
]][[]][[
][
IM
o
K
I
K
S
EE
][][
1][
IM
o
K
I
K
S
E
E
][][
1
][
E + S ES E + P
k1
+
I
EI E + Q
k3
KI
k-1
k2
k-3](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-90-320.jpg)
![91
][
][
2 ESk
dt
Pd
MK
SE
k
dt
Pd ]][[][
2
][
][][
1
][ 2
S
K
I
K
S
E
K
k
dt
Pd
IM
o
M
][
][
1
][][ 2
S
K
I
K
SEk
dt
Pd
v
I
M
o
oEkv 2max
1/v
[S]
1/vmax
[I]
][
][
1
][max
S
K
I
K
Sv
v
I
M
][
1][
1
11
maxmax SK
I
v
K
vv I
M
I
M
K
I
v
K ][
1
max
Inhibitor is replaced
from the active sites
by substrate at high [S]](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-91-320.jpg)

![93
E + S ES E + P
k1
+
I
+
I
EI + S EIS E + P
k3
KI KI
k-1
k2
k-3
][2 ESkv
MIIM
M
KK
SI
K
I
K
S
K
S
v
v
]][[][][
1
][
max
IM
M
K
I
S
K
S
K
S
v
v
][
1][
][
1
][
max
Vmax=k2E0
1/v
1/[S]
1/vmax-app
M
M
K
S
K
][
1
Iapp Kv
I
vv maxmaxmax
][11
1/vmax-app
[I]
1/vmax
1/(vmaxKI)
Henri-Michaelis-Menton Eq](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-93-320.jpg)

![95
][2 ESkv
MIM
M
KK
SI
K
S
K
S
v
v
]][[][
1
][
max
I
M
K
I
vSv
K
v
][
1
1
][
11
maxmax
Vmax=k2E0
1/v
1/[S]
1/v’
[I]
E + S ES E + P
KM
+
I
EIS E + Q
KI
IK
I
v
][
1
1
max
maxv
KM
IK
I
vv
][1
'
1
max
1/vmax
1/KI
Uncompetitive Inhibition](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-95-320.jpg)
![96
PESnSE n
n
n
SE
ES
K
]][[
][
n
n
n
n
SEKE
SEK
ESE
ES
Y
]][[][
]][[
][][
][
n
SK
Y
Y
][
1
KSn
Y
Y
log]log[
1
log
][2 ESkv
][][ ESEEo
n
o SEKEE ]][[][
n
o
SK
E
E
][1
][
n
no
SK
SKE
ES
][1
][
][
n
n
o
n
SK
SKEk
ESkv
][1
][
][ 2
2
maxmax ][
11
v
K
Svv n
Yield
Hill eq.
n: Hill coefficient](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-96-320.jpg)
![Chain Reactiosn
98
][
][
''1
]][['][
2
22
2
1
Br
HBr
k
BrHk
dt
HBrd
Chain reactions usually involve free radicals
The experimental rate law is
H2+Br2=2HBr](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-98-320.jpg)





![The Lindemann Mechanism
104
N2O5 = NO2 + NO3.
k1 >> k-2 [N2O5]
Rate k2[N2O5]2
k1 << k-2 [N2O5]](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-104-320.jpg)










![Langmuir-Hinshelwood
]][[
;
;
;
BSASkR
kSPBSAS
KBSSB
KASSA
s
s
B
A
2
)1( BBAA
BBAA
s
PbPb
PbPb
kR
A
B
s
AA
BB
s
AA
BBAA
s
P
P
k
Pb
Pb
k
Pb
PbPb
kR '
2
)(
2
)( BBAA
BBAA
s
PbPb
PbPb
kR
B
A
s
BB
AA
s
BB
BBAA
s
P
P
k
Pb
Pb
k
Pb
PbPb
kR '
)()( 2
BAs
BBAA
s PPk
PbPb
kR ,
1
115
bBPB <bAPA
bBPB > bAPA
bBPB , bAPA <1](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-115-320.jpg)
![Eley-Ridel Equation
skSCASB
KASSA
;
; 1
B
AA
AA
ss P
Pb
Pb
kASbkR
1
]][[
BsB
A
AA
sB
AA
AA
s PkP
P
Pb
kP
Pb
Pb
kR '
1
BAsBAAsB
AA
AB
s PPkPPbkP
Pb
Pb
kR '
1
116](https://image.slidesharecdn.com/chemicalkinetics-171007151323/85/Chemical-kinetics-116-320.jpg)



































































